






線粒體自噬抑制衰老過程中胞質mtDNA依賴的cGAS/STING炎癥激活
巨自噬隨著年齡的增長而減少,這一變化被認為是衰老過程的標志。線粒體自噬,一種線粒體必要選擇性自噬降解,是否也會隨著年齡的增長而減少,目前尚不清楚。在作者對Mito-QC報告小鼠多器官有絲分裂的分析中,老年小鼠的有絲分裂要么增加,要么不變。轉錄組學分析顯示,老年小鼠視網膜中I型干擾素反應明顯上調,與胞質mtDNA水平升高和CGAS/STING通路激活有關。重要的是,這種一致的改變在來自老年供體的原始人類成纖維細胞中不斷重復。在老年小鼠中,用尿石素A誘導線粒體自噬會降低CGAS/STING的激活并改善神經功能的惡化。這些發現表明,線粒體自噬的誘導是減少年齡相關炎癥和增加健康壽命的策略。該研究于2024年1月發表在《nature communications》,IF 16.6。
技術路線: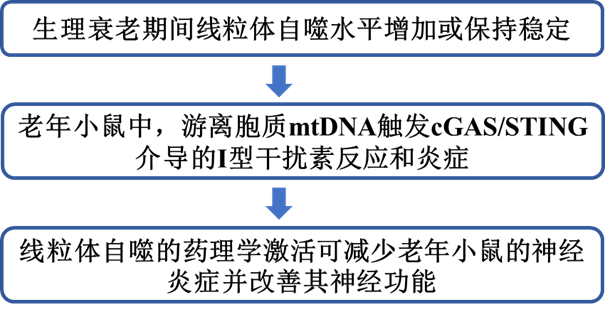
主要研究結果:
1. 生理衰老期間線粒體自噬水平增加或保持穩定
與衰老相關的線粒體自噬變化已被廣泛研究。然而,線粒體自噬在衰老組織中是上調還是下調仍不清楚。這在一定程度上是由于目前可用于線粒體自噬研究的工具和讀數的稀缺性和復雜性。在本研究中,作者使用可固定的mito-QC報告基因C57BL/6J小鼠評估了線粒體自噬,該小鼠普遍表達由pH不敏感的mCherry組成的嵌合蛋白,pH不敏感的GFP,以及線粒體分裂1蛋白(FIS1)的線粒體靶向序列(MTS)(圖1a)。因此,顯微鏡可用于區分細胞質線粒體(mCherry+GFP+)和經歷溶酶體降解的線粒體(mCherry+GFP?)。對年輕(6-8個月)和老年(24-26個月)小鼠組織樣本的共聚焦成像分析顯示,老年小鼠的腎臟、大腦、視網膜色素上皮(RPE)、神經視網膜、小腦和肝臟中的線粒體自噬水平顯著較高(圖1b)。相反,胰腺、脾臟、肌肉、心臟和肺部的線粒體自噬水平在各組之間沒有差異(圖1b)。先前的文獻報道表明,溶酶體降解不足會導致使用串聯熒光報告器對實驗的混淆解釋。由于溶酶體功能缺陷與衰老有關,作者評估了用蛋白酶抑制劑Leupeptin(40 mg/kg,16小時)或載體(鹽水)處理的年輕和老年小鼠視網膜中的線粒體溶酶體水平。
在所分析的大多數器官中觀察到PINK1誘導的泛素Ser65磷酸化增加(圖1c,d),表明與年齡相關的線粒體自噬增加是由PINK1/Parkin通路驅動的,該通路對線粒體膜電位的擾亂作出反應。作者還測量了視網膜中參與受體介導的線粒體自噬的蛋白質水平(BNIP3L/NIX、BNIP3、FKBP8、PHB2、FUNDC1),沒有發現與年齡相關的變化。
與作者和其他人的先前報告一致,與年輕小鼠相比,老年小鼠表現出總體巨自噬減少,如自噬體數量(LC3+)增加所證明的,而不伴隨自溶小體數量(LC3+LAMP1+)的變化而變化。這些在老年小鼠中的觀察結果,加上自噬銜接子Sequestome-1/p62和多泛素化蛋白的積累,以及自噬調節因子缺乏轉錄變化,表明巨自噬流量的普遍阻斷與線粒體自噬的增加相一致。由于線粒體功能障礙也被認為是衰老的主要驅動因素之一,作者試圖確定伴隨生理衰老的轉變的原因和后果,從而使線粒體的降解優于其他自噬底物。
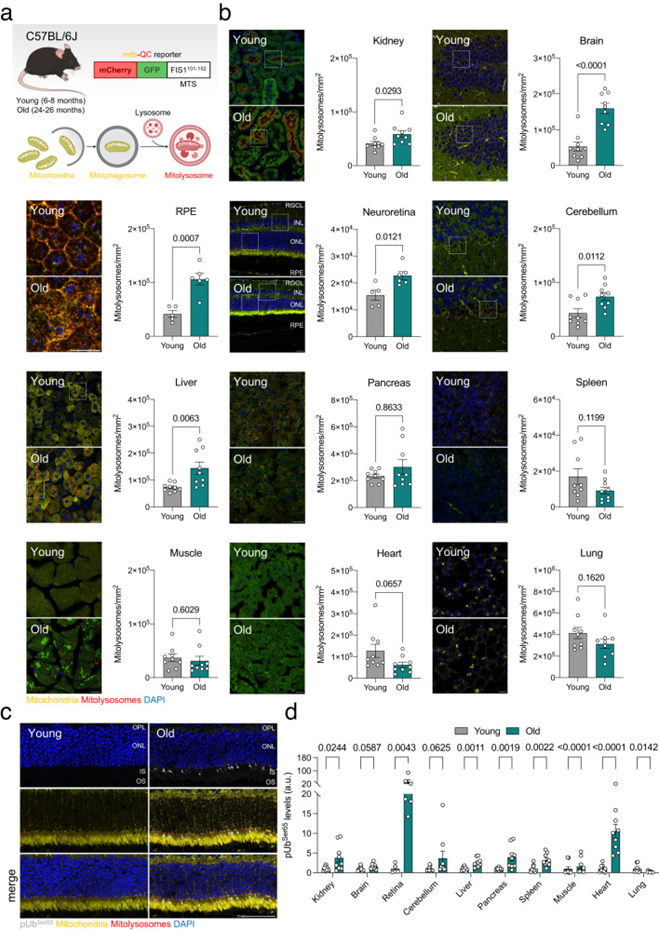
圖1:小鼠生理性衰老與多個器官的線粒體自噬穩定或增加有關
2. 老年小鼠中,游離胞質mtDNA觸發cGAS/STING介導的I型干擾素反應和炎癥
視網膜是中樞神經系統中一個定義明確、易于接近的部分,它是顯示出與年齡相關的線粒體自噬最強烈增加的器官之一。來自老年小鼠的視網膜顯示出與炎癥和對病毒或內源性胞質DNA有反應的幾種有關途徑上調(圖2a)。事實上,前三個陽性富集標志是干擾素反應(INFα,IFNγ)和TNFα信號傳導(圖2b)。使用干擾素進行更精細的分析顯示,67.5%的顯著上調的差異表達基因(DEG)對應于干擾素刺激的基因(ISG),并且所有基因都參與I型干擾素反應(圖2c)。考慮到視網膜的免疫特權性質和本研究中所有小鼠所接受的嚴格獸醫監測,作者假設內源性胞質DNA可能會觸發這種炎癥反應。年輕和老年視網膜的抗-DNA免疫染色顯示,老年組的胞質DNA量增加,其中大部分位于富含線粒體的區域(圖2d)。視網膜樣品的亞細胞分級用于進一步探索老年小鼠胞質DNA的增加,顯示老年小鼠與年輕小鼠相比,胞質部分的完整mtDNA水平(mt-Nd2、mt-Co1、mt-Cytb)顯著增加10倍(圖2e)。
干擾素基因(cGAS/STING)軸的環狀GMP-AMP合酶刺激因子已被確定為mtDNA釋放引發炎癥的主要傳感器和下游效應物之一。與轉錄數據一致,作者觀察到cGAS和STING的蛋白質水平顯著增加(圖2f),導致下游轉錄因子干擾素調節因子3(IRF3)的激活,如磷酸化-IRF3Ser396水平的增加所反映的(圖2f)。IRF3下游幾個基因(Ifit1、Ifi44、Ifih1、Cxcl10、Rtp4)的轉錄水平在老年視網膜中也顯著上調(圖2g)。因為在多個器官中觀察到與年齡相關的線粒體自噬增加(圖1b),作者還評估了肝、腦、腎和肌肉中的cGAS/STING激活。與年輕小鼠相比,老年小鼠的腎臟和肌肉樣本中的STING水平顯著較高。為證實這些發現,作者使用來自C57BL/6J小鼠的公開轉錄組數據(GSE141252)分析了17個不同器官和5個不同年齡組(4、9、12、18、24個月)的cGAS/STING介質和下游靶基因的水平。在所分析的17個器官中的12個(腎上腺、棕色脂肪組織、小腦、大腦額葉皮層、心臟、腎臟、大腸、肝臟、肺、肌肉、皮膚和白色脂肪組織)中觀察到轉錄水平增加的常見模式,表明老年小鼠中cGAS/STING介導的炎癥的增加不僅限于視網膜。
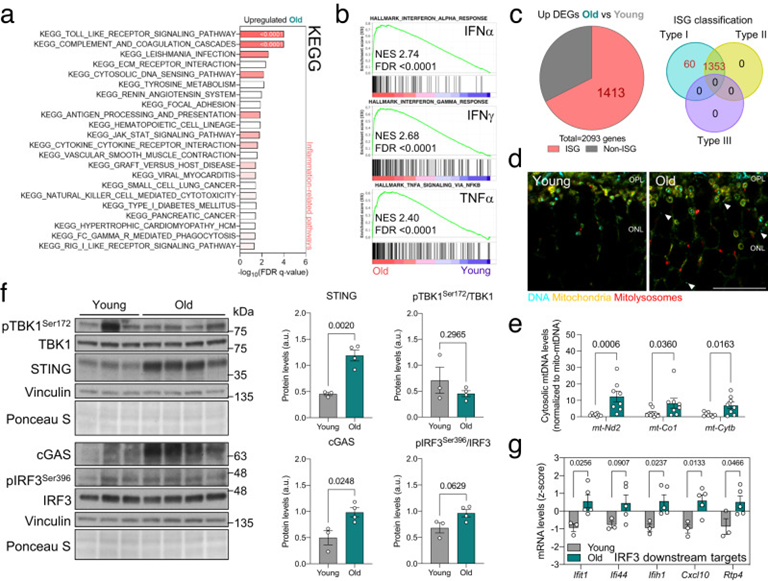
圖2:老年小鼠mtDNA釋放增加引發cGAS/STING介導的炎癥
最后,作者研究了在老年小鼠中觀察到的線粒體自噬、mtDNA釋放和cGAS/STING激活的增加是否也在不同物種中保持不變。首先,作者對先前發表的數據(GSE113957)進行轉錄組學分析,這些數據來自原代正常人真皮成纖維細胞(NHDF),根據臨床指南分為兒科、青年人、成人、中年人、老年人或老年人(圖3a)。與作者在小鼠中的發現相一致,老年組顯示出最高水平的cGAS/STING介質和下游IRF3靶點(圖3b),以及與所有其他組相比干擾素反應(INFα、IFNγ)和TNFα信號的富集(圖3b)。此外,與所有其他基因相比,在老年組上調的DEG中,50.3%對應于ISG,并被歸類為干擾素I型應答基因(圖3d)。IFIH1和RTP4(IRF3的下游靶標)表達的相關性分析顯示與年齡呈正相關(圖3e)。最后,來自年輕供體(28歲)和年老供體(62歲)(圖3f,g)的NHDF s in-house分析顯示后一組磷酸化-泛素化Ser65和胞質DNA焦點的顯著高水平(圖3h)。與在老年小鼠視網膜中觀察到的表型相似,在受體介導的線粒體自噬效應物的水平上沒有觀察到變化,也沒有觀察到心磷脂向線粒體外膜(OMM)的易位。這些數據支持cGAS/STING介導的炎癥增加和線粒體自噬之間的年齡依賴性關聯,并表明這種關聯在器官和物種中是保守的。這些觀察結果表明,PINK1/Parkin介導的線粒體自噬可能隨著衰老的進展而選擇性上調,以改善線粒體質量控制并抵消mtDNA的釋放,從而限制cGAS/STING的激活。
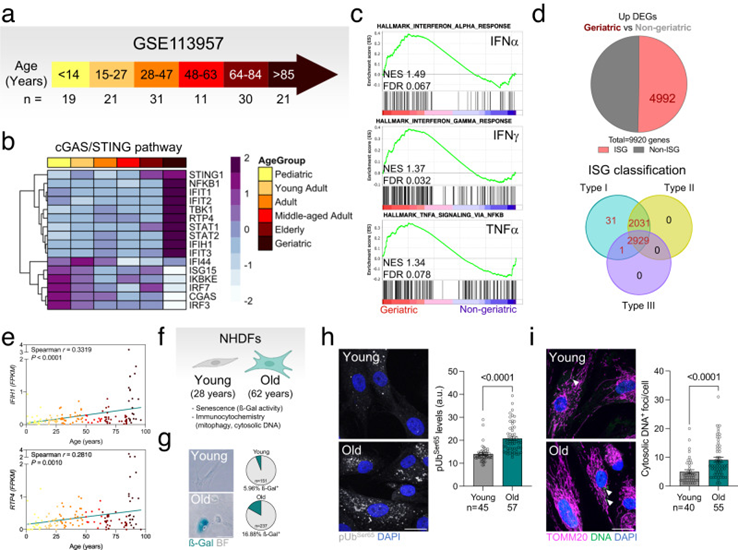
圖3:在老年供體的原代人真皮成纖維細胞中也觀察到cGAS/STING被細胞質DNA激活
3. 線粒體自噬的藥理學激活可減少老年小鼠的神經炎癥并改善其神經功能
最近的研究強調了線粒體自噬誘導劑在動物模型中延長壽命和健康壽命的能力,以及它們作為治療阿爾茨海默病和肌萎縮等年齡相關疾病的潛力。基于作者的觀察結果,即線粒體自噬隨著生理衰老而上調,作者假設通過藥物誘導線粒體自噬可以改善與衰老相關的一些有害變化(即神經炎癥以及神經和視覺功能下降)。作者使用線粒體自噬誘導劑尿鋰蛋白A(UA)進行了一項干預研究,尿鋰蛋白是一種源自石榴或樹莓等可食用植物和水果的天然代謝產物,已在臨床試驗中進行研究。作者對每天腹膜內注射UA(2.3 mg/kg)或載體8周的年輕(6個月)和老年(22個月)小鼠進行了神經生理學、行為學和視網膜免疫組織化學和轉錄組學分析(圖4a)。灌注腦的質譜(UPLC-ESI-QTOF-MS)和作為陽性對照的血漿樣品顯示,游離UA穿過血腦屏障并到達中樞神經系統(圖4a)。由于灌注的大腦樣本沒有經過酶水解,這是第一項明確顯示到達大腦的精確泌尿代謝形式的研究。因此,這些結果支持了先前的假設,即游離UA作為直接效應物參與腦組織。UA治療顯著增加了年輕和老年小鼠神經視網膜中的線粒體自噬(圖4b)和RPE(圖4c),盡管后者的程度較低。與媒介物治療的小鼠相比,UA治療的老年小鼠后肢抱膝得分(用作一般神經功能的讀數)顯著較低(圖4d)。與載體處理的小鼠相比,使用新型物體識別(NOR)測試評估,用UA處理的老年小鼠也顯示出改善的識別記憶(圖4e)。Rod介導的黃昏和夜間視力隨著年齡的增長而惡化,嚴重影響老年人的生活質量。通過視網膜神經生理學的視網膜電圖(ERG)評估,UA治療的老年小鼠顯示出暗視(暗)視力改善,這表明Rod介導的視覺功能改善(圖4f,g),以及參與視黃醇代謝的基因表達增加。在用UA治療的年輕或老年小鼠中,未觀察到中視(混合)和明視(光)視力的差異。在用UA處理的老年小鼠中,桿狀光轉導蛋白視覺阻滯蛋白(光刺激異常整合的指標)的內化也減少。免疫組織化學表明,UA改善了老年小鼠的CtBP2+mGluR6+突觸完整性(圖4h),進一步表明UA促進了衰老過程中視覺功能的保存。UA還降低了脂質過氧化衍生的4-HNE+聚集體(圖4i),表明線粒體質量控制的改善也可能減少衰老視網膜中的氧化應激。光學相干斷層掃描(OCT)分析顯示沒有主要的形態計量學變化(圖4j),主要視網膜細胞類型的數量發生了變化。
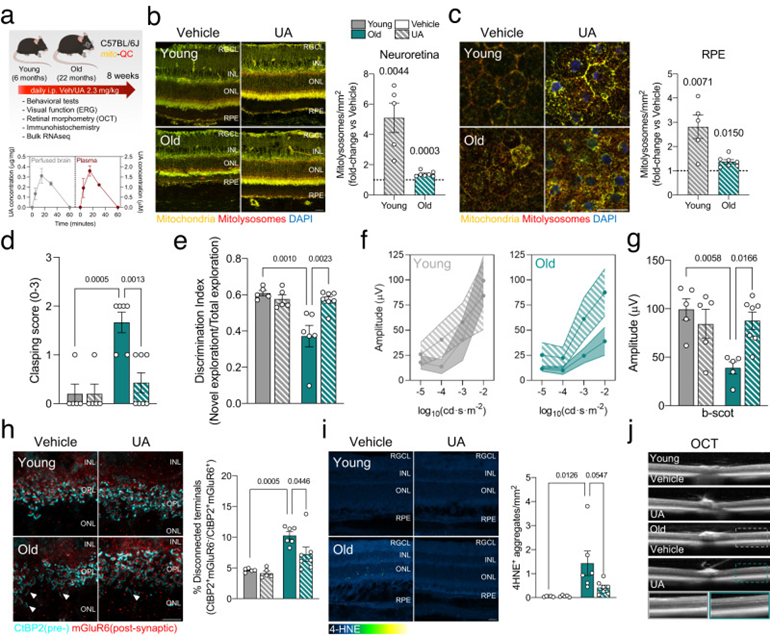
圖4:促進線粒體自噬可改善與年齡相關的神經衰退
至關重要的是,UA的線粒體自噬刺激降低了老年視網膜中胞質DNA的水平(圖5a)和DNA結合的cGAS(圖5a)。此外,干擾素反應(INFα、IFNγ)和TNFα信號傳導是老年小鼠中前3個陽性富集特征(圖2b),也是UA治療的老年小鼠中的前3個陰性富集特征(圖5b)。在用UA誘導線粒體自噬后,老年小鼠的cGAS/STING介質和下游基因的轉錄水平也降低(圖5c)。視網膜老化傳統上也與神經膠質炎癥反應增加有關。UA處理的老年小鼠顯示出膠質纖維酸性蛋白/GGFP+星形膠質細胞增生顯著減少(圖5d),并且有輕微的電離鈣結合銜接子分子1/Iba1+小膠質細胞浸潤減少的趨勢(圖5e),表明cGAS/STING信號減少也可以直接減少膠質細胞激活。
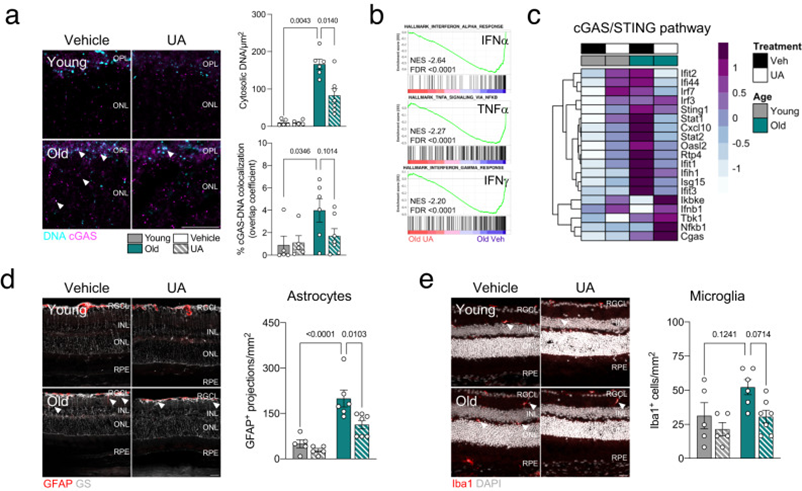
圖5:在老年小鼠中觀察到,UA治療可減弱cGAS/STING反應的增加
UA也被描述為同時刺激線粒體生物發生,以便在線粒體自噬誘導后恢復健康的線粒體。在UA治療的小鼠視網膜中,細胞質線粒體質量(mitoQC;FIS1-GFP+)確實增加,這一現象在老年小鼠中更為明顯(圖6a)。為進一步剖析cGAS/STING、線粒體自噬和線粒體穩態之間的相互作用,作者使用ARPE-19細胞系建立了線粒體DNA釋放的體外模型。ABT-737是一種Bcl-2抑制劑,它可以觸發線粒體外膜中Bax/Bak孔的形成,導致其內部成分(包括mtDNA18)的釋放。細胞也用泛胱天蛋白酶抑制劑Q-VDOPh(QVD)處理,以避免細胞色素c釋放引起的細胞凋亡誘導(圖6b)。用ABT-737處理增加了胞質DNA焦點的數量,當細胞與UA共同處理但不與選擇性cGAS抑制劑G140共同處理時,這種現象完全恢復(圖6c)。ABT-737也刺激了cGAS/STING信號級聯,UA和G140都消除了這種作用(圖6d)。總之,這些數據驗證了UA通過促進線粒體質量控制和消除其觸發事件(mtDNA釋放)而不是調節下游信號傳導來減少cGAS/STING介導的炎癥。值得注意的是,當細胞與G140共同處理時,ABT-737誘導的線粒體自噬減少,這表明線粒體自噬確實是對cGAS/STING激活的細胞保護反應(圖6e)。我們在老年C57BL/6J小鼠上(圖1c,d)和來自老年供體的NHDF(圖3h)重復了該發現,ABT-737顯著增加了磷酸化-泛素化Ser65水平,表明PINK1/Parkin依賴線粒體自噬的激活。類似地,在受體介導的線粒體自噬效應物中沒有觀察到變化、心磷脂水平或其向OMM的易位。模擬了作者在小鼠視網膜中的發現,UA還在ABT-737模型中誘導線粒體生物發生,并略微減少線粒體ROS的產生(圖6f)。最值得注意的是,線粒體呼吸測定分析顯示,UA恢復了ABT-737處理的細胞的基礎呼吸和ATP產生,并顯著提高備用呼吸能力(圖6g)。
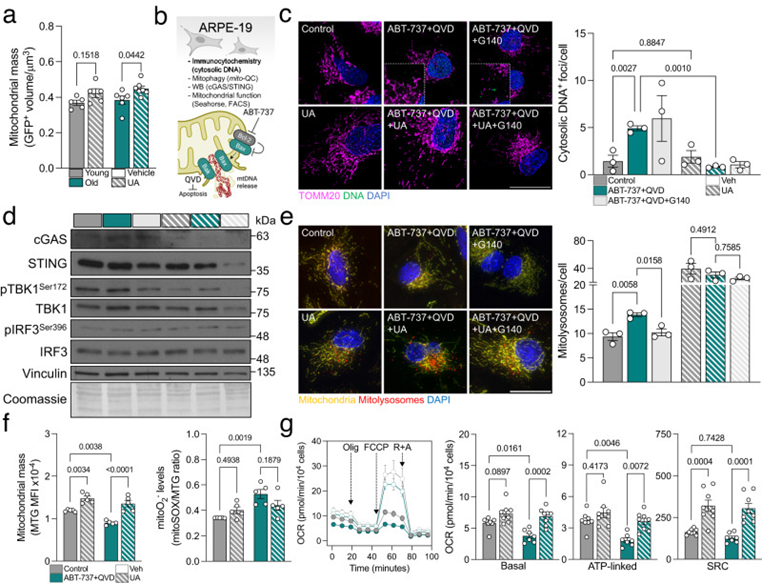
圖6:UA誘導線粒體生物發生,改善線粒體功能
最后,為完全確定線粒體自噬是否是UA衍生的有益作用的唯一原因,或者是否也涉及線粒體生物發生,作者在ARPE-19細胞中評估了相同的讀數,其中PINK1/Parkin依賴線粒體自噬(PINK1;PARK2 siRNA;圖7f)和/或線粒體生物發生(100μM氯霉素;圖7c)同時下調。PINK1/Parkin敲低有效地消除了ABT-737和UA的線粒體自噬誘導(圖7a,b)。線粒體生物發生抑制對線粒體自噬水平沒有影響(圖7a,b),也沒有UA誘導的胞質DNA減少(圖7d,e)。然而,在ABT-737處理的細胞中,PINK1/Parkin敲低完全消除了UA介導的胞質DNA焦點的減少(圖7e,f),表明該途徑確實是抑制mtDNA釋放的原因。
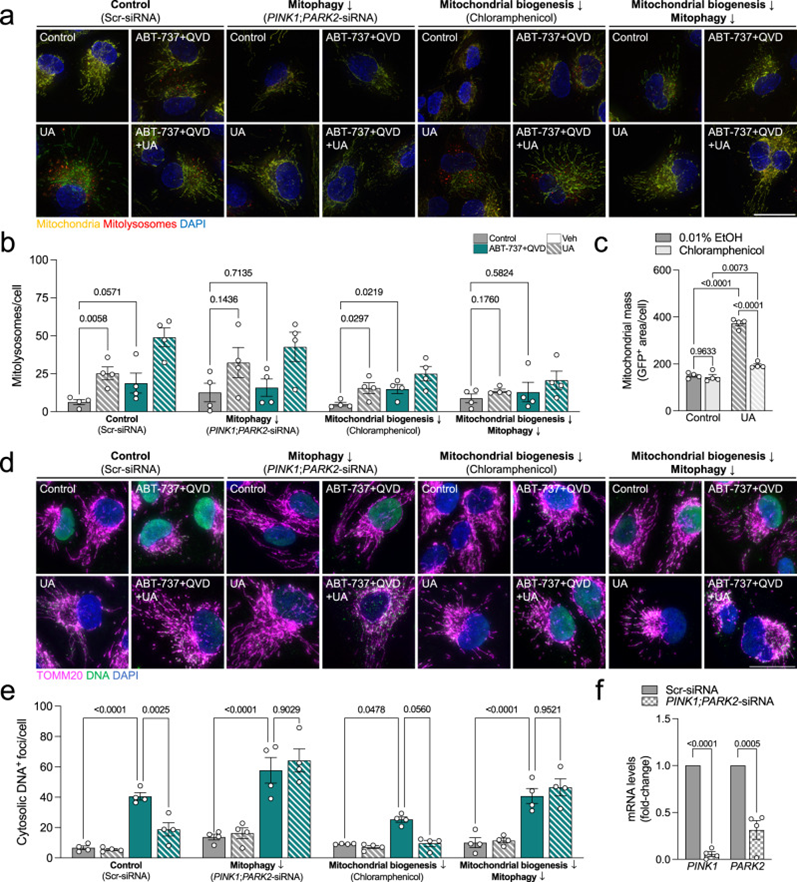
圖7:UA 的 PINK1/Parkin 依賴性線粒體自噬刺激介導胞質 ABT-737 誘導的 DNA 減少
結論:
總之,這些數據構成了支持藥物誘導線粒體自噬減輕年齡相關炎癥和神經系統惡化的治療潛力的概念證明。
實驗方法:
線粒體自噬動物模型構建,細胞培養,免疫熒光,qPCR,WB,尿石素代謝物測定,動物行為測試,透射電鏡,RNA測序,流式
參考文獻:
Jiménez-Loygorri JI, Villarejo-Zori B, Viedma-Poyatos á, Zapata-Mu?oz J, Benítez-Fernández R, Frutos-Lisón MD, Tomás-Barberán FA, Espín JC, Area-Gómez E, Gomez-Duran A, Boya P. Mitophagy curtails cytosolic mtDNA-dependent activation of cGAS/STING inflammation during aging. Nat Commun. 2024 Jan 27;15(1):830. doi: 10.1038/s41467-024-45044-1. Erratum in: Nat Commun. 2024 Feb 19;15(1):1504. PMID: 38280852; PMCID: PMC10821893.