






肝Zbtb18(含鋅指和BTB結構域的18)通過FXR(法尼醇X受體)減輕肝脂肪性肝炎
脂肪酸合成和消耗之間的持續失衡會導致非酒精性脂肪性肝病(NAFLD),以及肝炎和胰島素抵抗。然而,潛在機制的細節尚不完全清楚。在本研究中,作者揭示了轉錄因子Zbtb18在NAFLD患者和小鼠模型的肝臟中的表達顯著降低。肝臟Zbtb18敲除促進了NAFLD的特征,如能量消耗受損和脂肪酸氧化(FAO),并誘導胰島素抵抗。相反,在高脂飲食(HFD)喂養的小鼠或糖尿病小鼠中,肝臟Zbtb18過表達減輕了肝脂肪變性、胰島素抵抗和高血糖。值得注意的是,體外和體內機制研究表明,Zbtb18轉錄激活法尼醇X受體(FXR)介導的FAO和網格蛋白重鏈(CLTC)蛋白可抑制NLRP3炎癥小體的活性。通過在小鼠和培養的小鼠原代肝細胞(MPHs)中敲除和強制表達FXR,進一步驗證了肝細胞Zbtb18表達減輕NAFLD和隨后的肝纖維化的關鍵機制。此外,敲除CLTC顯著消除了肝臟Zbtb18過表達對巨噬細胞NLRP3炎癥小體活性的抑制作用。綜上所述,Zbtb18通過轉錄激活FXR介導的FAO和CLTC的表達,從而抑制NLRP3炎癥小體的活性,減輕炎癥應激和胰島素抵抗,為治療肝脂肪變性和纖維化提供了有價值的藥物。本文于2024年1月發表于《Signal Transduction and Targeted Therapy》,IF:39.3,Q1。
技術路線:

主要研究結果:
1、肝臟Zbtb18表達下調與NAFLD的發生發展密切相關
為了系統全面地識別參與脂肪性肝炎發生發展的關鍵信號分子,作者首先對NAFLD患者和正常對照的肝組織進行了mRNA微陣列分析,并隨后將該臨床轉錄組數據與已發表的轉錄組數據(來自NAFLD患者的GSE213621和來自NAFLD小鼠模型的GSE35961)進行了交叉分析,以確定差異表達基因(DEGs)。結果顯示,3個轉錄組分別有274、411和792個差異表達基因。此外,3個數據集中有33個DEGs重疊。值得注意的是,作者發現在這些轉錄組數據中,肝臟Zbtb18的表達下調(圖1a)。此外,作者證實了NAFLD患者肝活檢組織中Zbtb18 mRNA和蛋白的顯著降低(圖1b)。作者還發現在幾種NAFLD小鼠模型(包括db/db糖尿病小鼠、ob/ob肥胖小鼠和HFD誘導的肥胖小鼠)中,肝臟Zbtb18表達降低(圖1c-e)。此外,在油酸和棕櫚酸(OA和PA)培養的小鼠原代肝細胞(MPHs)中,肝臟Zbtb18的表達顯著降低(圖1f-g)。為了初步探究Zbtb18的生理功能,作者構建了AD -Zbtb18感染的MPHs,發現Zbtb18過表達有效地增加了參與FAO和氧化磷酸化(OXPHOS)的基因表達,而顯著抑制了參與糖異生的基因(圖1h, i)。因此,Zbtb18過表達增加了脂肪酸的分解代謝,同時減少了MPHs中的脂質沉積(圖1j-l)。因此,作者的研究表明肝臟Zbtb18與NAFLD的發生發展呈負相關。
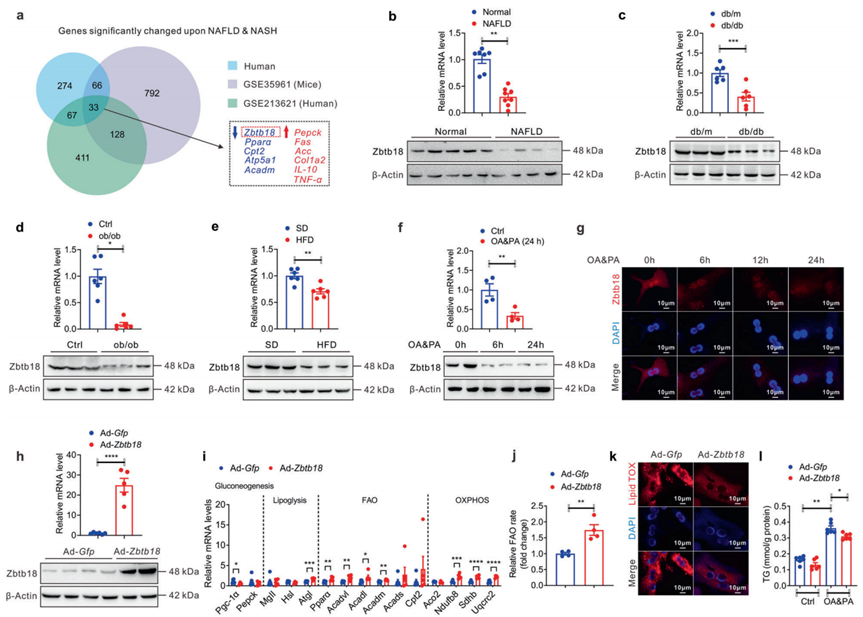
圖1 肝臟Zbtb18 mRNA和蛋白表達下調與NAFLD的發生發展密切相關
2、肝臟Zbtb18的消融使小鼠更容易發生脂肪性肝炎
為了探索肝臟Zbtb18的功能,作者首先生成了肝細胞特異性Zbtb18缺失(Zbtb18LKO)小鼠。以Zbtb18flox/flox小鼠作為Zbtb18LKO小鼠對照。接下來,作者長期給予這些小鼠高脂飲食(HFD),以進一步探索肝臟Zbtb18在脂質代謝中的生理作用。作者發現,肝臟Zbtb18缺失易使小鼠在長時間HFD暴露后發生肥胖,如小鼠較高的體重和脂肪量所示(圖2a, b)。組織學檢查(HE染色)也一致顯示Zbtb18LKO小鼠附睪白色脂肪組織(WAT)、腹股溝WAT和肩胛間棕色脂肪組織(BAT)的脂肪細胞肥大增加(圖2c)。相應地,這些小鼠肩胛間BAT中產熱基因的表達受到抑制,這可能導致了它們的體重增加(圖2d)。然后,作者將小鼠飼養在代謝籠中,發現與對照小鼠相比,Zbtb18LKO小鼠的能量消耗、呼吸O2消耗和CO2生成程度較低(圖2e)。更重要的是,Zbtb18LKO小鼠顯示HFD喂養后小鼠的空腹血糖和胰島素水平增加(圖2f)。此外,Zbtb18LKO小鼠表現出葡萄糖耐量和胰島素敏感性的損害(圖2g)。隨后的蛋白質印跡分析也顯示了Zbtb18LKO小鼠肝臟中AKT和GSK-3β磷酸化的降低,進一步表明這些小鼠的胰島素敏感性受損(圖2h)。此外,肝臟Zbtb18缺失降低了參與FAO和OXPHOS的基因,上調了糖原生成基因,共同增加了高脂飲食小鼠肝臟中的脂質沉積(圖2i, j)。
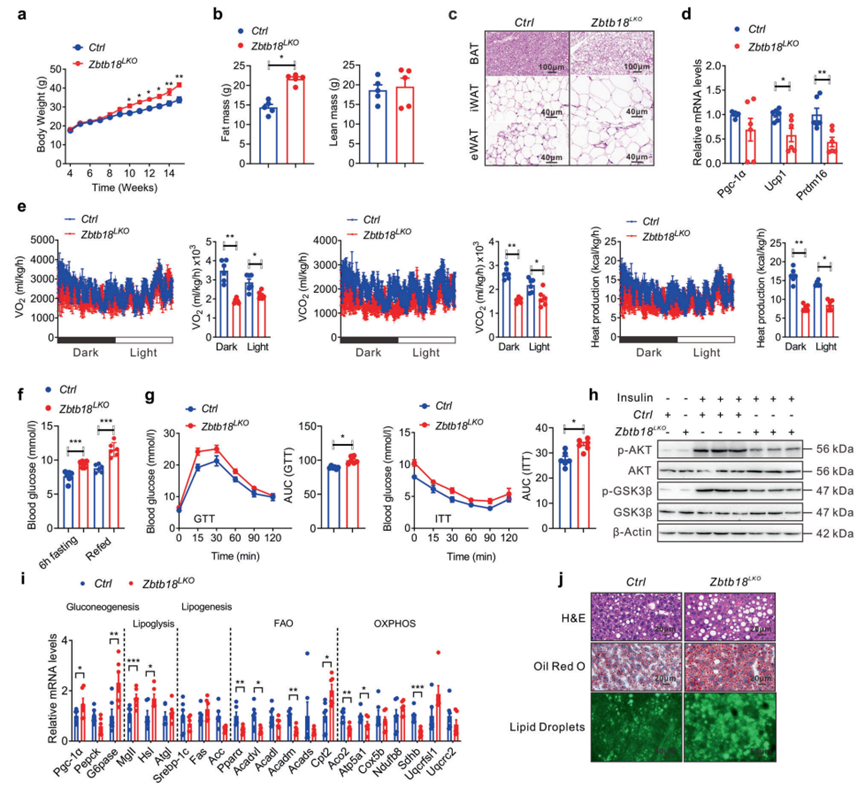
圖2 肝臟Zbtb18的消融加重了HFD喂養的小鼠的脂肪性肝炎
3、肝臟Zbtb18過表達對高脂飲食誘導的肝脂肪變性的保護作用
為了驗證肝臟Zbtb18的增加是否可以改善肝臟的脂質和葡萄糖穩態,作者通過將Alb-Cre小鼠和Rosa26-Zbtb18小鼠雜交,產生了肝臟特異性Zbtb18過表達(Zbtb18LKI)小鼠。以Zbtb18Rosa26基因敲入小鼠作為Zbtb18LKI對照。作者首先將這些小鼠暴露于HFD,發現Zbtb18過表達降低了體重,這可能歸因于脂肪量和肝臟重量的減少(圖3a, b)。此外,與同窩對照小鼠相比,Zbtb18LKI小鼠表現出能量消耗、呼吸氧消耗和CO2生成的增加,以及BAT中Ucp1(解偶聯蛋白1)基因表達的增加,最終導致體重下降(圖3c, d)。值得注意的是,Zbtb18過表達有效緩解了HFD誘導的肝脂肪變性,包括肝細胞氣球樣變和脂質沉積的改善(圖3e)。此外,Zbtb18LKI小鼠的血清酮體水平升高,肝臟和血清TGs水平降低(圖3f, g)。相應地,Zbtb18過表達增加了參與脂肪分解、FAO和OXPHOS的基因表達,而與脂肪生成相關的基因表達略有變化(圖3h)。此外,改善肝臟過度脂質蓄積可有效減輕HFD誘導的肝臟脂毒性。這減輕了炎癥細胞的浸潤和炎癥細胞因子的釋放,最終逆轉了HFD引起的肝炎,改善了肝功能,表現為炎癥基因表達和血清ALT、AST水平的降低(圖3i-l)。同樣,由于肝臟Zbtb18的過表達,這些小鼠表現出較低的空腹血糖和胰島素水平(圖3m, n)。肝臟Zbtb18過表達還改善了HFD喂養的小鼠的葡萄糖耐量和胰島素敏感性,并提高了肝臟中AKT和GSK-3β的磷酸化,表明HFD暴露導致的胰島素信號通路改變得到了恢復(圖3o, p)。綜上所述,這些數據證明了肝臟Zbtb18在維持全身糖脂平衡中發揮著重要作用。
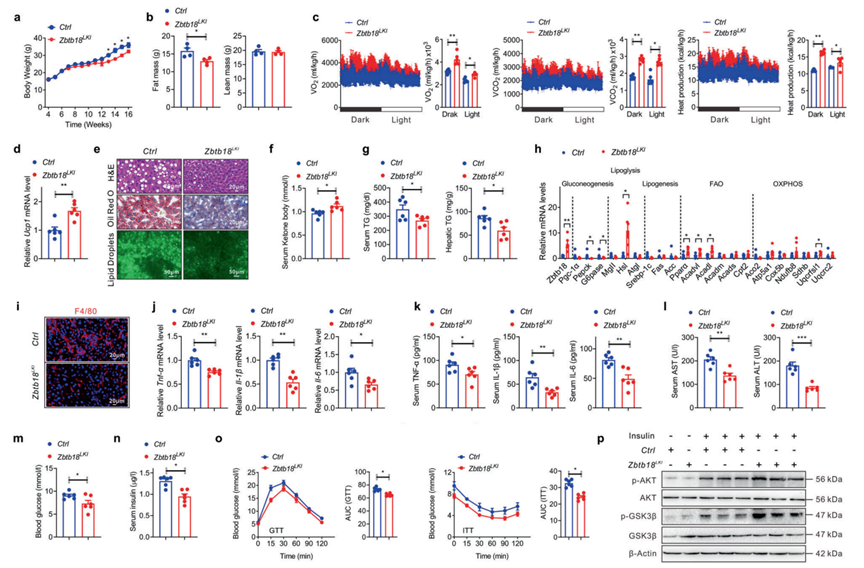
圖3 肝臟Zbtb18過表達對抗高脂飲食誘導的肝脂肪變性
4、強制肝臟Zbtb18表達減輕糖尿病小鼠的肝脂肪變性
鑒于肝臟Zbtb18在保護飲食誘導的糖脂紊亂方面的主要作用,作者接下來測試其過表達是否會改善糖尿病db/db小鼠的脂肪肝表型。因此,通過尾靜脈注射AAV-Zbtb18, db/db小鼠的肝臟Zbtb18表達增加(補充圖7)。與此一致,AAV-Zbtb18感染的db/db小鼠體重和肝臟重量/體重比值降低(圖4a, b)。AAV介導的肝臟Zbtb18過表達也顯著改善了db/db小鼠的脂肪肝表型,如TGs含量的總體變化和組織學分析所示(圖4c, d)。同樣,AAV-Zbtb18感染的db/db小鼠顯示空腹血糖和胰島素水平降低(圖4e)。后一結果暗示db/db小鼠中葡萄糖紊亂的改善,這通過改善的葡萄糖耐受不良和胰島素抵抗進一步證實(圖4f)。此外,在注射AAV-Zbtb18的db/db小鼠中也觀察到血清酮體水平升高(圖4g)。相應地,肝臟Zbtb18過表達顯著增加了脂肪酸氧化相關基因的表達,而糖原基因包括Pgc-1α和Pepck被抑制(圖4h)。同時,肝臟中AKT和GSK-3β磷酸化的增加促進了db/db小鼠胰島素功能障礙的恢復(圖4i)。同時,Zbtb18過表達的db/db小鼠肝臟F4/80和cd11b陽性巨噬細胞在肝臟內的聚集顯著減少,同時炎性細胞因子水平降低(圖4j, k)。所有這些變化都有助于肝功能障礙的改善,正如這些小鼠血清ALT和AST水平的降低所證明的(圖4l)。總之,這些數據表明,靶向肝臟Zbtb18是阻止db/db小鼠肝臟脂肪變性進展的有效方法。
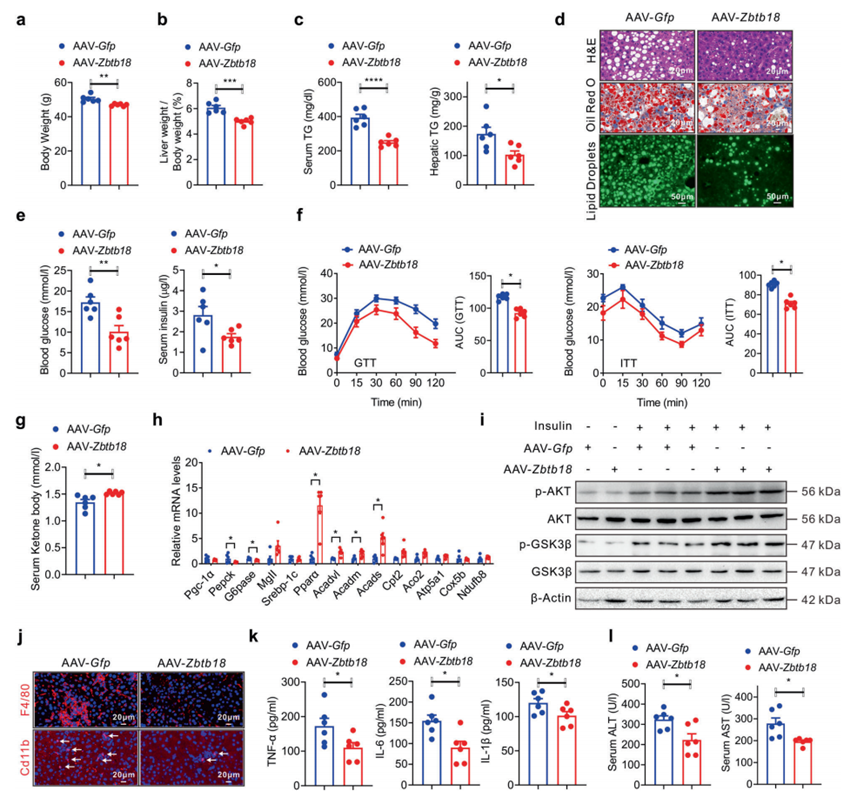
圖4 挽救肝臟Zbtb18表達可減輕糖尿病小鼠的肝脂肪變性
5、Zbtb18通過轉錄激活FXR介導的FAO加速脂質分解代謝
為了研究在Zbtb18介導的肝臟保護作用中涉及的效應器或信號節點的細節,作者對Zbtb18過表達的MPHs進行了RNA-seq。因此,作者發現7347個基因因Zbtb18的過表達而發生了不同的變化,其中3628個基因上調,3719個基因下調(圖5a)。隨后的GO分析表明,Zbtb18過表達伴隨著肝臟炎癥和脂肪酸代謝通路的明顯顯著改變(圖5b)。為了深入了解Zbtb18調節肝臟脂肪酸代謝和炎癥的潛在機制,作者分析了從Zbtb18過表達的MPHs和NAFLD患者肝臟中發現的重疊DEGs。值得注意的是,結果顯示大部分重疊的DEGs涉及FXR靶基因(圖5c, d)。此外,在Zbtb18過表達的MPHs和NAFLD患者的肝臟中鑒定的上述DEGs的基因集富集分析(GSEA)顯示,“FXR信號通路”與Zbtb18的表達呈正相關,提示這些DEGs的表達可能是由FXR依賴的機制驅動的(圖5e)。此外,在Zbtb18過表達的肝臟中,FXR mRNA和蛋白水平均上調(圖5f-g)。與此一致,Zbtb18過表達也顯著增加了培養的MPHs中FXR蛋白及其靶基因的總水平和細胞核水平(圖5h)。
為了進一步探索Zbtb18激活FXR的潛在機制,作者在MPHs中進行了Zbtb18特異性ChIP-seq。結果顯示在FXR基因中有一個顯著的Zbtb18結合啟動子區域(圖5i)。此外,作者進一步分析了ChIP-seq數據,并證實位于FXR啟動子區域的“AACTCACTCT”是Zbtb18的關鍵結合基序(圖5j)。然后,使用一系列已報道的構建體(pFXR-1838, pFXR-1074和pFXR-677)在HepG2和MIHA細胞中進行熒光素酶檢測。作者發現,Zbtb18顯著刺激pFXR-1838和pFXR-1074的轉錄活性,而pFXR-677沒有產生影響,這表明Zbtb18轉錄因子的潛在結合位點從-1074 bp到-677 bp(圖5k, l,左)。更重要的是,FXR啟動子區AACTCACTT突變為TTTTTTTT減弱了Zbtb18對FXR轉錄活性的刺激作用,表明其結合位點位于FXR區域(圖5k, l,右)。此外,作者的ChIP-qPCR分析顯示,在db/db小鼠和HFD喂養的小鼠的肝臟中,FXR啟動子上的Zbtb18轉錄因子的占用顯著降低,這進一步詳細說明了這些小鼠中FXR失調的機制(圖5 m)。為了進一步驗證FXR在介導Zbtb18誘導的保護作用中的關鍵作用,作者構建了FXR敲除的MPHs,并發現FXR缺失顯著削弱了Zbtb18對FAO的保護作用,導致MPHs中的脂質積累水平未發生改變(圖5n-p)。相反,FXR過表達減輕了Zbtb18缺乏引起的MPHs中脂質沉積(圖5q)。綜上所述,這些數據表明Zbtb18介導的FXR轉錄激活與肝細胞脂質穩態之間存在很強的相關性。
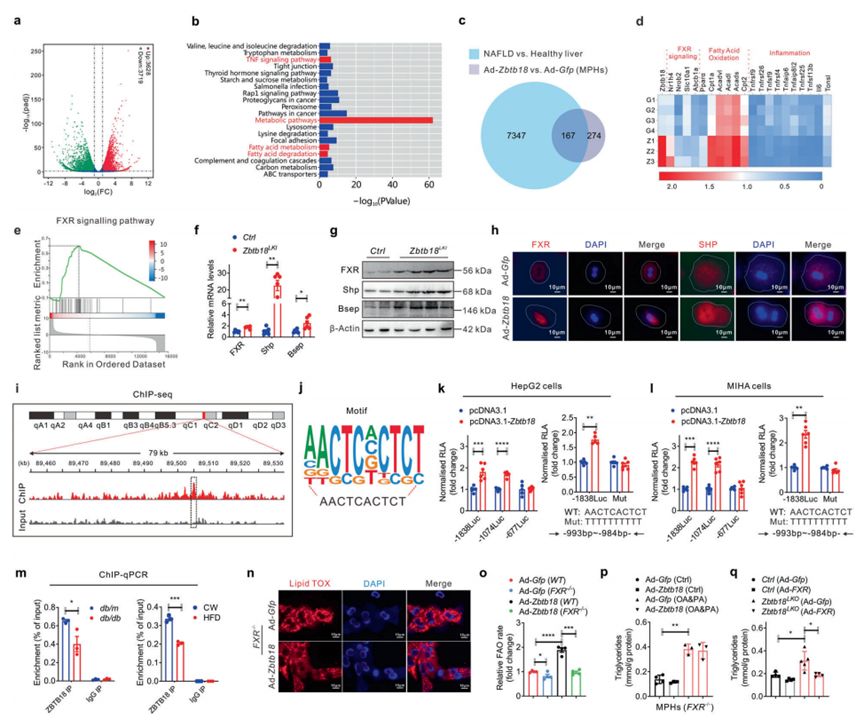
圖5 FXR的Zbtb18轉錄激活通過FAO加速脂質分解代謝
6、肝臟Zbtb18通過FXR介導的CLTC蛋白表達抑制巨噬細胞中NLRP3炎癥小體的活化
越來越多的證據表明,肝細胞脂質失衡通常會引發并促進肝臟炎癥,進而加重NAFLD的進展。為了全面探討Zbtb18對NAFLD發展的影響,作者還研究了NLRP3炎癥小體的組裝和活化的變化,在肝臟Zbtb18獲得或丟失模型中,NLRP3炎癥小體在肝臟炎癥中起關鍵作用。肝臟Zbtb18缺失顯著增強了HFD誘導的炎癥小體相關蛋白的表達,包括NLRP3, ASC和Caspase-1。這些作用被肝臟Zbtb18過表達顯著逆轉(圖6a, b)。作者的結果表明,肝臟Zbtb18對NLRP3介導的炎癥小體激活具有抑制作用,這也在AAV-Zbtb18感染的db/db小鼠中得到了驗證(圖6c)。隨后,作者對Ad-Zbtb18感染的肝細胞上清液進行了蛋白質組學分析,以揭示參與肝細胞Zbtb18驅動的炎癥抑制的關鍵遞質。隨后對使用上述培養基處理的巨噬細胞進行的實驗也證實了這一假設,實驗顯示在Ad-Zbtb18感染的肝細胞條件培養基中培養的巨噬細胞中,炎癥小體的活性受到抑制(圖6d)。有趣的是,在Zbtb18缺失的小鼠中,FXR缺失有效地消除了Zbtb18對NLRP3炎癥小體相關蛋白的抑制,而FXR過表達有效地阻斷了NLRP3炎癥小體的異常激活(圖6e-f)。在肝細胞中敲除CLTC可有效減弱Zbtb18條件培養基體外誘導的巨噬細胞中NLRP3炎癥小體的抑制作用(圖6g-h)。綜上所述,這些結果表明肝臟Zbtb18的表達通過FXR介導的CLTC蛋白表達抑制巨噬細胞中NLRP3炎癥小體的活化,從而幫助減輕小鼠NAFLD。
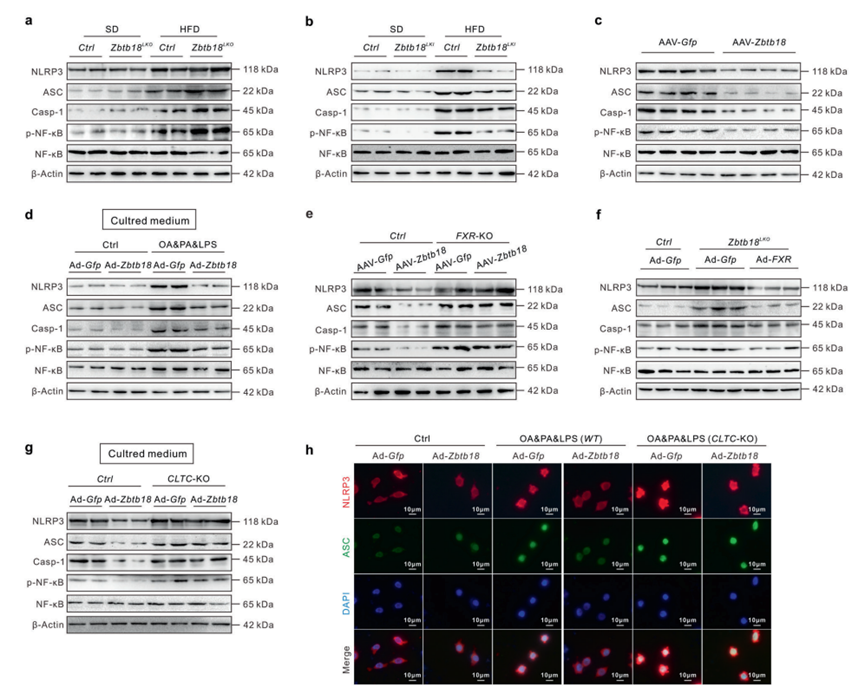
圖6 肝臟Zbtb18蛋白通過FXR介導的CLTC蛋白表達抑制巨噬細胞NLRP3炎癥小體的活化
7、FXR活性有效地介導了Zbtb18誘導的肝脂肪變性的減輕
與上述結果一致,作者通過尾靜脈注射AAV-Zbtb18到FXR缺失的小鼠體內來測試FXR在介導Zbtb18驅動的有益效應中的生理功能。因此,作者發現FXR缺失幾乎消除了Zbtb18誘導的體重和脂肪量減少,HFD小鼠的能量消耗發生了罕見的變化(圖7a, b)。此外,注射AAV-Zbtb18后,FXR缺失小鼠的血清酮體水平幾乎沒有變化(圖7c)。此外,肝臟Zbtb18過表達未能改變與FAO相關的肝臟基因的表達(圖7d)。這導致AAV-Zbtb18感染的FXR敲除小鼠的血清和肝臟中的TGs含量不變,同時肝細胞內的氣球樣變和脂滴積聚不變(圖7e, f)。同樣,FXR缺失消除了Zbtb18過表達后空腹血糖和血清胰島素水平的下降(圖7g)。此外,AAV-Zbtb18感染的FXR缺陷小鼠的葡萄糖耐量和胰島素抵抗無變化(圖7h)。這些結果表明,FXR在Zbtb18誘導的肝臟葡萄糖紊亂保護作用中起關鍵作用。值得注意的是,在FXR缺失的小鼠中沒有觀察到Zbtb18誘導的肝臟中AKT和GSK-3β磷酸化的增加(圖7i)。此外,在AAV-Zbtb18感染小鼠和AAV-Gfp感染小鼠之間,由于FXR缺失,未檢測到肝臟生糖基因(如Pgc-1α和Pepck)的表達有差異(圖7j)。Zbtb18同樣不能降低FXR缺陷小鼠的血清炎性細胞因子,如TNF-α和IL-1β,以及ALT和AST水平(圖7k -1)。
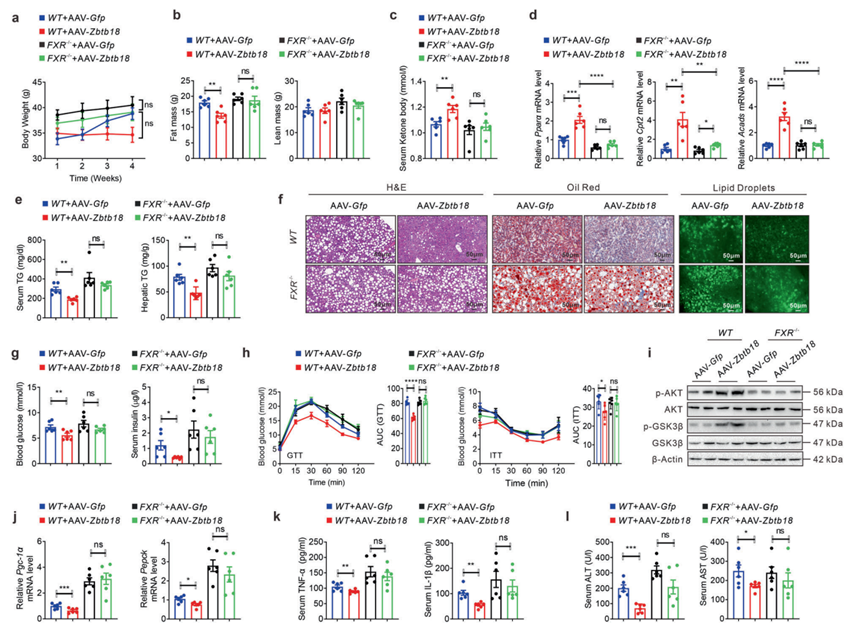
圖7 FXR消融減弱了Zbtb18誘導的對脂肪性肝炎的保護作用。
相反,FXR的強制表達有效地增加了其靶基因在肝Zbtb18LKO小鼠肝臟中的表達(圖8a)。由于FXR的表達,這些小鼠的脂肪量減少,能量消耗增加(圖8b, c)。相應地,Zbtb18LKO小鼠肝臟重量/體重比值、血清酮體水平和TGs含量的變化由于FXR的恢復激活而逆轉,同時這些小鼠中涉及fao的肝臟基因表達和肝臟脂肪變性的改善,如肝細胞內脂滴積聚導致的氣球樣變減少(圖8d-f)。此外,FXR表達增加降低了Zbtb18LKO小鼠的空腹血糖和胰島素水平。此外,FXR表達的增加顯著減輕了Zbtb18缺失引起的肝臟葡萄糖耐受不良和胰島素抵抗(圖8g, h)。在FXR過表達后,作者觀察到對肝臟中AKT和GSK-3β磷酸化的類似刺激作用,這表明Zbtb18LKO小鼠的肝臟胰島素信號活性得到挽救,并且參與糖生成的基因得到有益的調節(圖8i, j)。值得注意的是,強制肝臟FXR表達有效減輕了Zbtb18缺陷刺激的CD11b+和F4/80+巨噬細胞在肝臟中的聚集,從而減輕了炎癥應激,最終促進了這些小鼠NAFLD的改善(圖8k-m)。綜上所述,這些數據強烈表明了Zbtb18介導的FXR轉錄激活在維持肝臟糖脂穩態方面的潛在工作機制。
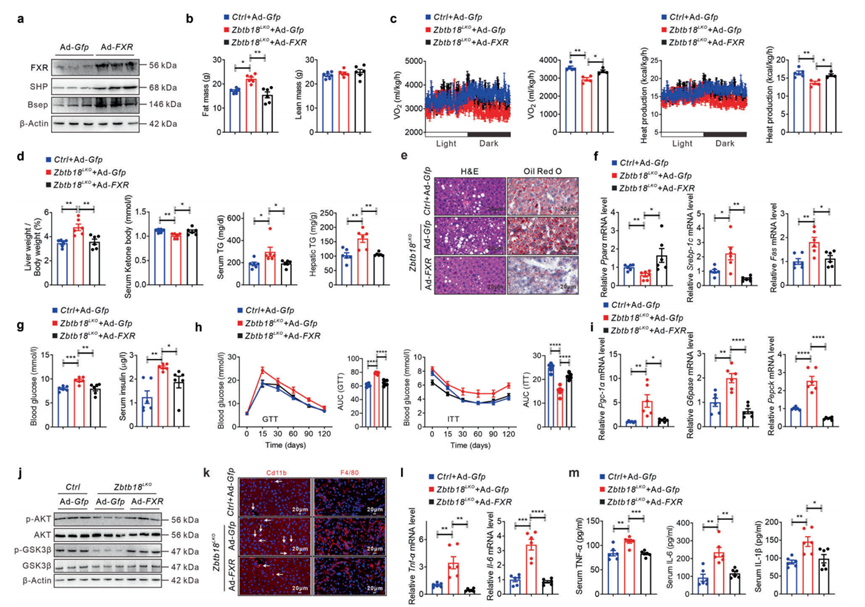
圖8 肝臟FXR強制表達減輕肝臟Zbtb18缺失小鼠的NAFLD表型
8、肝臟Zbtb18蛋白通過轉錄激活FXR保護MCD誘導的小鼠肝纖維化
越來越多的證據表明,肝臟內脂質的長期異常積累會導致纖維結締組織的過度沉積以及肝臟內細胞外基質的合成和降解失衡,從而促進NASH進展。考慮到Zbtb18在減少肝臟脂質沉積方面的明顯作用,作者懷疑Zbtb18可能對肝纖維化具有顯著的保護作用。接下來,作者用MCD飲食喂養Zbtb18LKO小鼠,以證明Zbtb18在MCD飲食誘導的肝纖維化發展中的生理功能。正如預期的那樣,與同窩對照小鼠不同,在MCD飲食暴露后,Zbtb18LKO小鼠的ALT和AST水平升高(圖9a),這表明這些小鼠發生了重度肝損傷和肝細胞死亡。此外,在MCD飲食喂養后,Zbtb18缺失導致TGs水平和肝臟脂質沉積均加重,如脂滴積累和隨后的肝細胞氣球樣變所示(圖9b, c)。此外,作者的天狼星紅染色分析表明,在Zbtb18消融的肝臟小鼠中,MCD飲食誘導的致死性實質內細胞外周纖維化的發生促進了α-SMA染色水平的急性升高(圖9d)。相應的,MCD飲食誘導的與肝纖維化發展相關的肝基因表達(包括α-SMA、Col1a1和TGF-β)也在肝Zbtb18LKO小鼠中受到刺激,同時FXR及其靶基因的表達受損(圖9e、f)。此外,肝臟Zbtb18缺失加劇了MCD飲食誘導的肝臟炎癥應激,如炎癥基因(如F4/80和TNF-α)的上調,血清炎癥細胞因子水平的升高,以及肝臟中F4/80+和CD11b+巨噬細胞的聚集增加(圖9e, g, h)。所有這些事件都會導致脂肪性肝炎的快速發展。
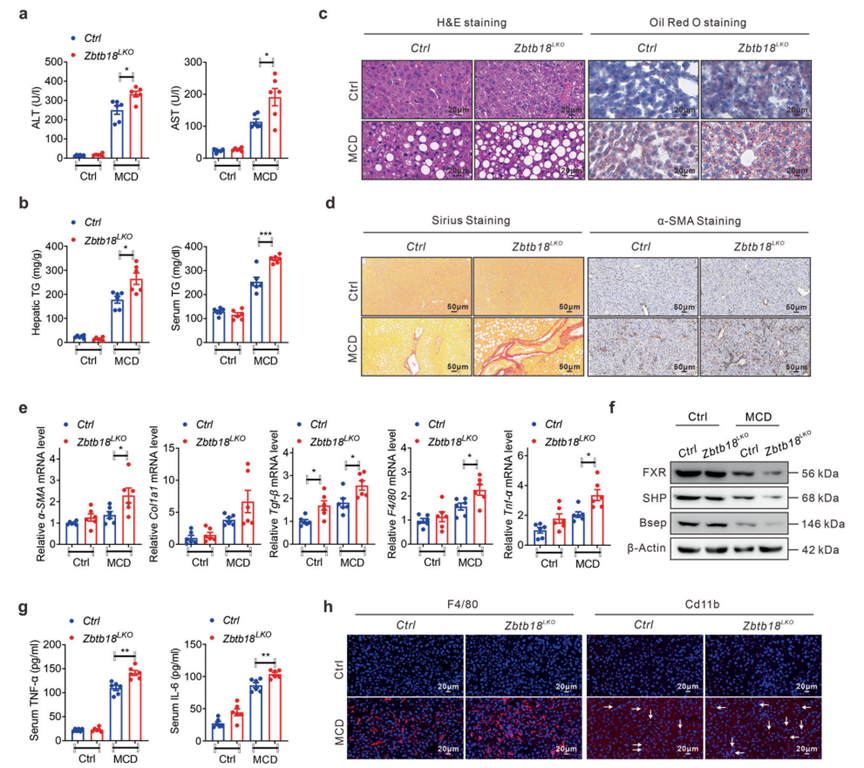
圖9 肝臟Zbtb18缺失加重MCD誘導的肝纖維化進展
相比之下,如正常血清ALT和AST水平與同窩對照組小鼠相比所示,肝臟Zbtb18過表達保護小鼠免受MCD飲食誘導的肝纖維化(圖10a)。此外,通過HE染色和油紅O染色分析顯示,Zbtb18過表達有效改善了MCD飲食刺激的TGs升高和肝臟脂肪沉積(圖10b, c)。Sirius染色和α-SMA免疫組化分析一致表明,在肝臟Zbtb18過表達的小鼠中,即使喂食MCD飲食,實質內細胞外周纖維化的發展也受到了部分阻礙(圖10d)。與此同時,密切相關的基因(包括α-SMA、Col1a1和TGF-β)表達減少(圖10e)。此外,肝臟Zbtb18過表達抑制了MCD飲食誘導的肝臟炎癥基因F4/80、TNF-α、CXCL1和CXCL10,從而降低炎癥細胞因子的血清水平和肝臟炎癥細胞的浸潤,有助于改善脂肪性肝炎(圖10e-g)。
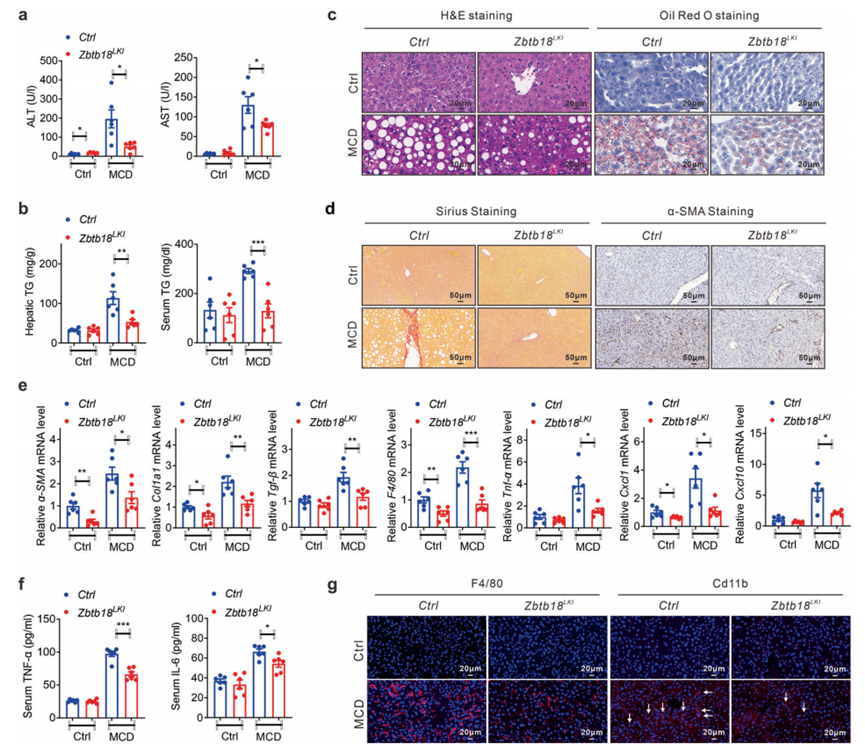
圖10 肝臟Zbtb18過表達對MCD誘導的小鼠肝纖維化具有保護作用
總之,這些發現提示肝臟Zbtb18在防御脂肪性肝炎方面具有潛在的治療作用,其部分機制是通過激活FXR及其下游靶基因(圖11)。
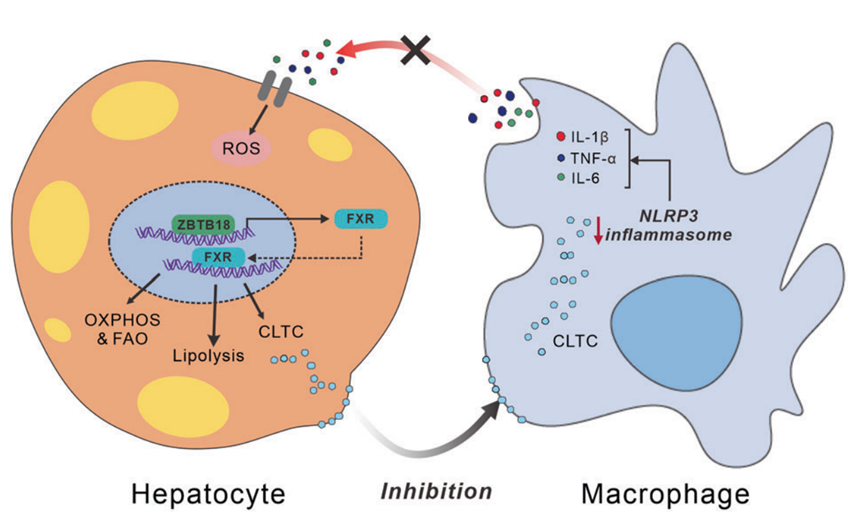
圖11 Zbtb18通過轉錄激活FXR介導的肝臟脂質代謝,抑制NLRP3炎癥小體的活性,減輕炎癥應激和胰島素抵抗
結論:
綜上所述,作者證明了肝臟Zbtb18可以通過直接結合FXR啟動子區的AACTCTCT元件來增加FXR mRNA和蛋白的表達。反過來,FXR可以刺激其靶基因加速FAO,從而阻止NAFLD的發生和發展。此外,Zbtb18/FXR軸刺激的CLTC蛋白顯著減輕肝臟炎癥浸潤和纖維化。因此,Zbtb18/FXR軸是一個新的治療NAFLD和NASH的候選靶點。
實驗方法:
臨床組織制備;動物和治療;組織學分析、免疫熒光分析和脂滴染色;脂肪酸氧化檢測;耐量試驗;代謝籠和MRI;ELISA和TGs和TCs檢測;熒光素酶報告基因法;染色質免疫沉淀(ChIP)分析;RNA測序;CHIP測序和發現峰;Western blot;實時定量PCR;細胞培養和預處理。
參考文獻:
Zhang L, Chen J, Yang X, Shen C, Huang J, Zhang D, Liu N, Liu C, Zhong Y, Chen Y, Tang K, Guo J, Cui T, Duan S, Li J, Huang S, Pan H, Zhang H, Tang X, Chang Y, Gao Y. Hepatic Zbtb18 (Zinc Finger and BTB Domain Containing 18) alleviates hepatic steatohepatitis via FXR (Farnesoid X Receptor). Signal Transduct Target Ther. 2024 Jan 24;9(1):20. doi: 10.1038/s41392-023-01727-7. PMID: 38263084; PMCID: PMC10806020.