






急性缺血誘導空間和轉錄上不同的小膠質細胞亞群
缺血性卒中后,缺血核心和梗死灶周圍區的損傷影響患者預后。微膠質細胞立即對缺血性打擊做出反應,引發免疫炎癥,在卒中后的細胞損傷中發揮著重要作用。然而,微膠質細胞的多樣性和涉及的機制仍然不清楚。我們首先對三個時間點的大鼠大腦進行了scRNA-seq和空間轉錄組學(ST),以確定與卒中相關的微膠質細胞亞簇及其空間分布。此外,通過RNAscope和免疫熒光技術在大鼠中驗證了微膠質細胞亞群特異性標記基因的表達和不同微膠質細胞亞群的定位。進行基因集變異分析(GSVA)以揭示微膠質細胞亞群的功能特征。此外,使用調查通路分析(IPA)來探索微膠質細胞亞群的上游調控因子,通過免疫熒光、RT-qPCR、shRNA介導的基因敲除和定向代謝組學進行確認。最后,在特定微膠質細胞亞群操縱后,評估大鼠MCAO模型中的梗死灶大小、神經功能缺陷和神經元凋亡。我們在MCAO大鼠大腦中發現了與卒中相關的微膠質細胞亞群。我們還確定了這些微膠質細胞亞群的新標記基因,并根據它們的空間分布將這些細胞定義為缺血性核心相關(ICAM)和梗死灶相關(IPAM)的微膠質細胞。ICAM由損傷相關分子模式誘導,可能由糖酵解提供動力,并表現出增加的促炎細胞因子和趨化因子產生。BACH1是驅動ICAM生成的關鍵轉錄因子。相反,梗死灶中富含的糖皮質激素可能觸發IPAM的形成,這些細胞可能由檸檬酸循環和氧化磷酸化提供動力,并以中等的促炎反應、抗炎代謝特征和髓鞘營養特性為特征。ICAM可能引起過度神經炎癥,加劇腦損傷,而IPAM可能表現出神經保護特性,對于梗死灶中細胞的穩態和存活可能至關重要。我們的研究結果為以特定微膠質細胞亞群為靶點的治療策略提供了生物學基礎,成為缺血性卒中的潛在治療策略。該研究于2023年12月發表在《Genome medicine》,IF:12.3。
技術路線:
結果:
1、細胞類型的鑒定
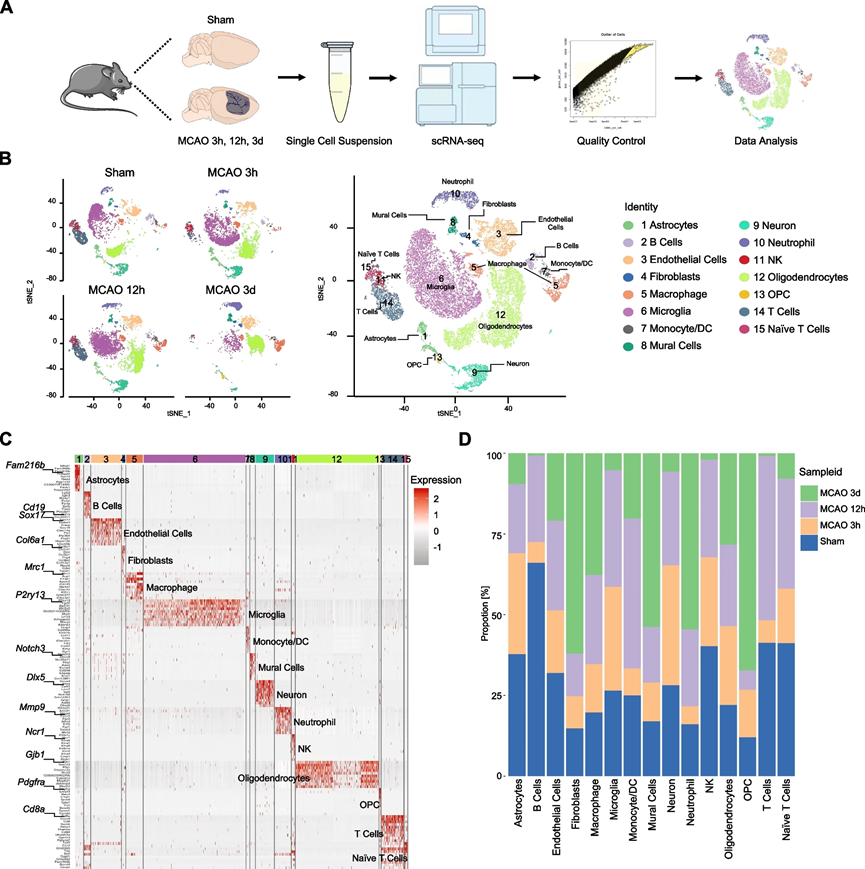
為了全面表征缺血性卒中后缺血半球主要細胞類型的變化,我們在經歷短暫性大鼠中腦動脈阻塞(MCAO)的大鼠的腦樣本中,采用scRNA-seq技術,在再灌注后的不同時間點(3小時、12小時和72小時)以及模擬大鼠上進行了細胞分離(圖1A)。3小時和12小時的時間點代表了缺血性卒中的亞急性階段,而72小時的時間點被選為進行急性階段變化的轉錄分析。總共評估了39,333個細胞,每個細胞平均含有1672-8800個獨特的分子標識符(UMIs),經過嚴格的CellRanger質量控制。在四個組中,每個細胞的平均基因數為901-2636。
接下來,我們進行了無監督聚類,并根據經典基因標記表達進行了細胞類型的注釋。如t-分布隨機鄰居嵌入(t-SNE)圖中所示,鑒定了15種不同的細胞類型,包括B細胞、內皮細胞、壁細胞、自然殺傷細胞(NK細胞)、單核/樹突細胞(DC細胞)、成熟型寡能神經膠質細胞(OPC)、T細胞、星形膠質細胞、成纖維細胞、巨噬細胞、微膠質細胞、神經元、中性粒細胞、寡能神經膠質細胞和原始T細胞(圖1B)。值得注意的是,微膠質細胞、寡能神經膠質細胞和內皮細胞占細胞總數的大多數。我們確定了每種細胞類型的所有標記基因,并在熱圖中顯示了前10個標記基因(圖1C)。
與先前的發現一致,缺血性打擊后浸潤的巨噬細胞和中性粒細胞數量逐漸增加,表明血腦屏障功能失調。此外,OPC的數量在手術后72小時顯著增加,表明中風后OPC的增殖,這與先前的研究結果一致。我們還觀察到在缺血后星形膠質細胞數量逐漸減少,而在72小時時微膠質細胞顯著減少,這可能歸因于急性缺血性損傷(圖1D)。總體而言,這些數據提供了對缺血半球細胞組成的基本描述。
2、缺血誘導兩種類型的小膠質細胞亞簇
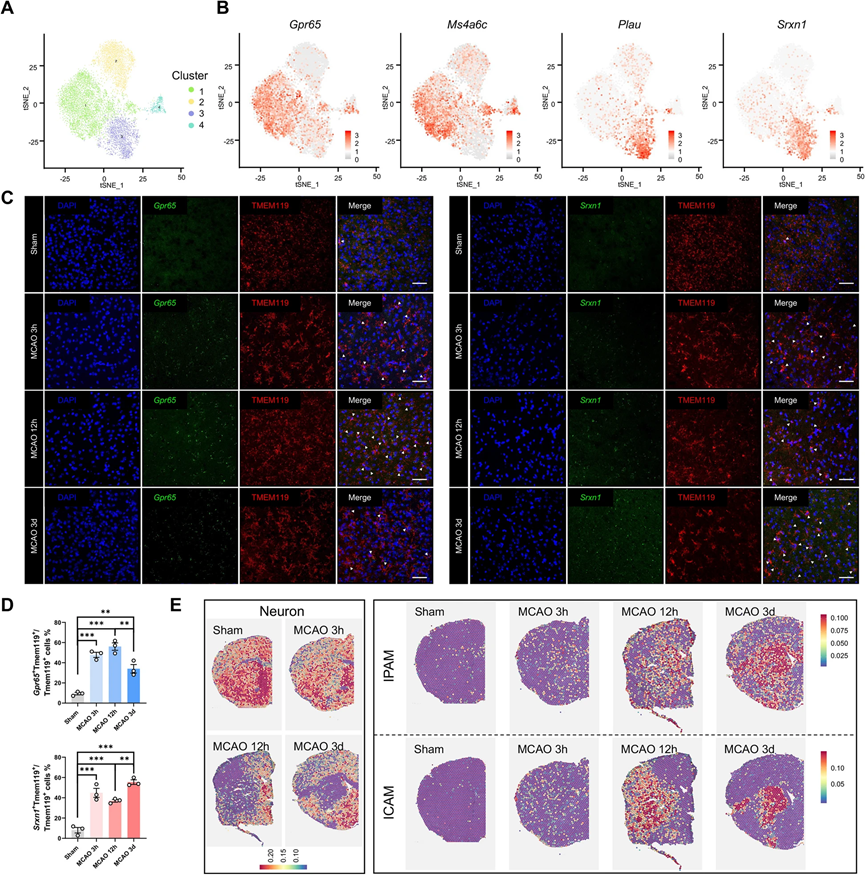
考慮到微膠質細胞在大腦中的重要性和微膠質細胞轉錄組的高質量,我們將焦點放在了單細胞水平的微膠質細胞上。我們在不同時間點確定了微膠質細胞的差異表達基因(DEGs)。與先前的研究結果一致,微膠質細胞DEGs的KEGG分析表明,在缺血性卒中的急性階段,微膠質細胞的吞噬和炎癥能力增強,代謝發生改變。此外,微膠質細胞被分為四個亞群進行進一步的分析(圖2A)。根據亞群比例圖,模擬組中94%的微膠質細胞屬于亞群2。此外,微膠質細胞的穩態基因(Tmem119、P2ry12、P2ry13、Csf1r、Cx3cr1、Hexb)在亞群2中高度表達,表明亞群2可能由穩態微膠質細胞組成。亞群4(465個細胞)包括相對較少數量的微膠質細胞,在模擬和MCAO大鼠之間變化不大;因此,亞群4的表型和功能特征未進一步研究。亞群1和亞群3占據了MCAO組中微膠質細胞的大多數。這些發現將亞群1和亞群3定位為缺血性卒中急性階段的關鍵角色,并提出了它們如何協調缺血性攻擊適應的問題。
進行差異基因表達分析以識別亞群特異性標記基因。大多數高表達的基因也是亞群特異性的,表明微膠質細胞亞群具有獨特的基因表達模式(圖2B)。亞群1表達了先前未報道的標記基因(例如,Pik3ip1、Cd300lf、Gpr65、Ms4a6c、Cep152、Ddit4、Zbtb16、Abhd15、Bmf和Arhgap24)。在亞群3中,Lgals3、Plau、Srxn1、Ankrd33b、Gas2l3、Nes、Edn1、Fam129b、Vat1和Fmn1的表達上調。前10個亞群特異性標記基因可可靠地識別微膠質細胞亞群1和亞群3。此外,疾病相關微膠質細胞(DAM)曾被確定為與阿爾茨海默病(AD)相關的一種獨特的微膠質細胞亞型。我們檢查了我們數據集中已確定的DAM標記基因的表達水平。在四個亞群中,一些基因(例如Tyrobp、Trem2、Apoe、Timp2和B2m)的表達保持不變。在亞群3中,一些基因的表達上調,例如Lgal3、Fth1、Cstb、Csf1、Cd63、Cd9、Ccl3和Lilrb4a,而與穩態微膠質細胞(亞群2)相比,亞群1中這些基因的表達保持不變,表明亞群3可能表現出DAM樣的特征。
為了確定亞群1和亞群3是否具有時間特性,我們使用RNAscope(ACD,Newark,CA,USA)進行了RNA-蛋白共染色,使用亞群1特異性標記基因Gpr65和亞群3特異性標記基因Srxn1標記不同的微膠質細胞群體(圖2C)。在再灌注后,亞群1和亞群3均作為對缺血性打擊的迅速反應者(圖2D)。為了確認亞群1和亞群3是否也存在于其他關于缺血性卒中的單細胞RNA測序研究數據中,我們重新分析了幾個現有的缺血性卒中后微膠質細胞的數據集(GSE174574,GSE197731,GSE227651)。與我們的結果一致,亞群1和亞群3在它們的t-SNE微膠質細胞圖中可以清晰地區分出來。
為了確定亞群1和亞群3是否與梗死核心存在任何空間關系,我們使用了空間轉錄組學(ST)來研究模擬和MCAO大鼠的腦切片。盡管ST幾乎無法達到單細胞分辨率,但仍然可以通過計算與scRNA-seq數據相關的亞群特異性標記基因的平均表達來獲得腦切片中不同細胞亞群的大致位置。為了定義缺血性核心,我們還查詢了在腦切片上每個點的下調表明神經元喪失的神經元基因。引人注目的是,亞群3標記基因占據了缺血性核心,而亞群1標記基因則環繞著缺血病灶在缺血后12小時(圖2E)。因此,我們將亞群1定義為缺血性梗死后的微膠質細胞(IPAM),將亞群3定義為缺血性梗死核心相關的微膠質細胞(ICAM)。
為了驗證這些發現,我們在免疫熒光實驗中使用ICAM特異性的LGALS3作為ICAM標記,而選擇PIK3IP1作為IPAM標記。在12小時和72小時時,ICAM標記在缺血性核心中顯著富集。IPAM標記在12小時時在缺血性梗死灶周圍的位置也得到了確認。此外,ICAM特異性標記(Lgals3)和IPAM特異性標記(Pik3ip1、Gpr65和Ms4a6c)也在ST中可視化顯示。我們還根據圖2E將腦切片分為兩個區域(缺血性核心和梗死周圍區)。在缺血性核心中上調的基因被定義為缺血性核心基因(ICGs),而在梗死周圍上調的基因被稱為缺血性梗死周圍基因(IPGs)。與ICGs重疊的ICAM標記更多,與IPGs重疊的IPAM標記也更多,進一步驗證了ICAM和IPAM的空間分布。因此,我們的數據揭示了兩個在缺血性卒中應答中涉及的在空間上和轉錄上具有不同特性的微膠質細胞亞群。
3、ICAM 和 IPAM 表現出截然不同的特征
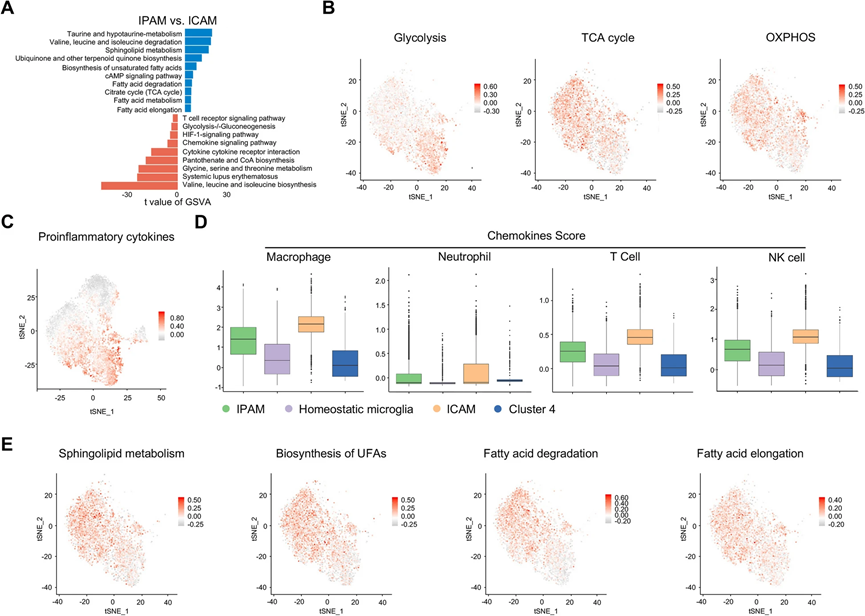
對于功能實驗,我們對IPAM和ICAM進行了基因集變異分析(GSVA)(圖3A)。我們發現ICAM可能在代謝上更依賴于糖酵解,而IPAM可能依賴于三羧酸循環(TCA)和氧化磷酸化(OXPHOS),通過使用Seurat中的AddModuleScore 計算與感興趣的代謝途徑相關的基因的表達來證實(圖3A、B)。一致地,“氧化磷酸化”和“檸檬酸循環”在IPG中也富集,如KEGG分析所示。ICAM中“斷裂”的TCA循環也可能導致中間產物水平的增加,如ICAM中增強的泛酮酸和輔酶A生物合成所示。此外,在IPAM中觀察到AMPK和cAMP信號通路的上調,表明IPAM使用了完全不同的能源來源(圖3A)。此外,微膠質細胞的代謝重編程依賴于HIF-1α通路,ICAM中HIF-1α通路也高度表達(圖3A)。
我們進一步發現相對于其他亞群,ICAM中存在一些促炎和炎癥響應基因(Il1a、Il1b、Il6、Il18、Tnf、Hmox1、Ptgs2)的表達上調(圖3C)。此外,巨噬細胞和中性粒細胞在MCAO后12小時開始浸潤大腦,并在72小時時聚集在梗死核心,這表明這些外周免疫細胞在空間上與ICAM存在重疊。由于微膠質細胞是缺血性卒中中各種趨化因子的主要來源,我們檢查了ICAM和IPAM中驅動不同外周免疫細胞招募的一系列趨化因子的表達。因此,ICAM中富集了不同外周免疫細胞中相關趨化因子基因集的表達(圖3D)。具體而言,與IPAM相比,ICAM中Ccl2、Ccl3、Ccl4、Ccl5、Cxcl2和Cxcl16的表達水平更高。此外,KEGG分析一致表明,促炎信號通路(“TNF信號通路”、“IL-17信號通路”、“MAPK信號通路”)在ICG中顯著富集。這些發現表明由ICAM介導的過度促炎和趨化反應,提示ICAM可能在缺血性卒中急性階段加速組織損傷,具有不利影響。
此外,與ICAM相比,GSVA分析顯示,IPAM中與氨基酸、脂質和碳水化合物相關的幾個代謝途徑顯著富集(圖3A)。支鏈氨基酸(BCAAs)如纈氨酸、亮氨酸和異亮氨酸的水平在ICAM中升高,而在IPAM中BCAAs降解被增強(圖3E)。此外,IPAM中其他氨基酸的代謝,如牛磺酸,也富集(圖3A)。根據GSVA數據,脂質代謝在IPAM中也很活躍,包括鞘脂代謝、主要膽酸生物合成、不飽和脂肪酸生物合成、脂肪酸降解和延長,通過AddModuleScore確認(圖3A,3E)。此外,IPAM中富集的代謝途徑(“BCAAs降解”、“不飽和脂肪酸生物合成”、“脂肪酸降解”、“脂肪酸延長”、“鞘脂代謝”)在ST中也顯示了一個梗死周圍的分布。
由于脂質代謝產物是髓鞘的重要組成部分,IPAM可能通過供應脂質組分來補償寡突細胞。此外,我們使用CellPhoneDB 分析了IPAM-寡突細胞相互作用中的配體-受體相互作用。已經確定了幾對配體-受體對,其中GRN-SORT1是其中最顯著的一對。為了測試GRN是否具有寡突細胞營養的潛力,我們用重組小鼠GRN(rmGRN)處理寡突細胞,發現rmGRN顯著促進了MAG的表達。
總的來說,這些獨特的特征至少在一定程度上塑造了微膠質細胞的應答,并為理解微膠質細胞亞群在對缺血刺激的響應中不同的激活狀態提供了新的視角。
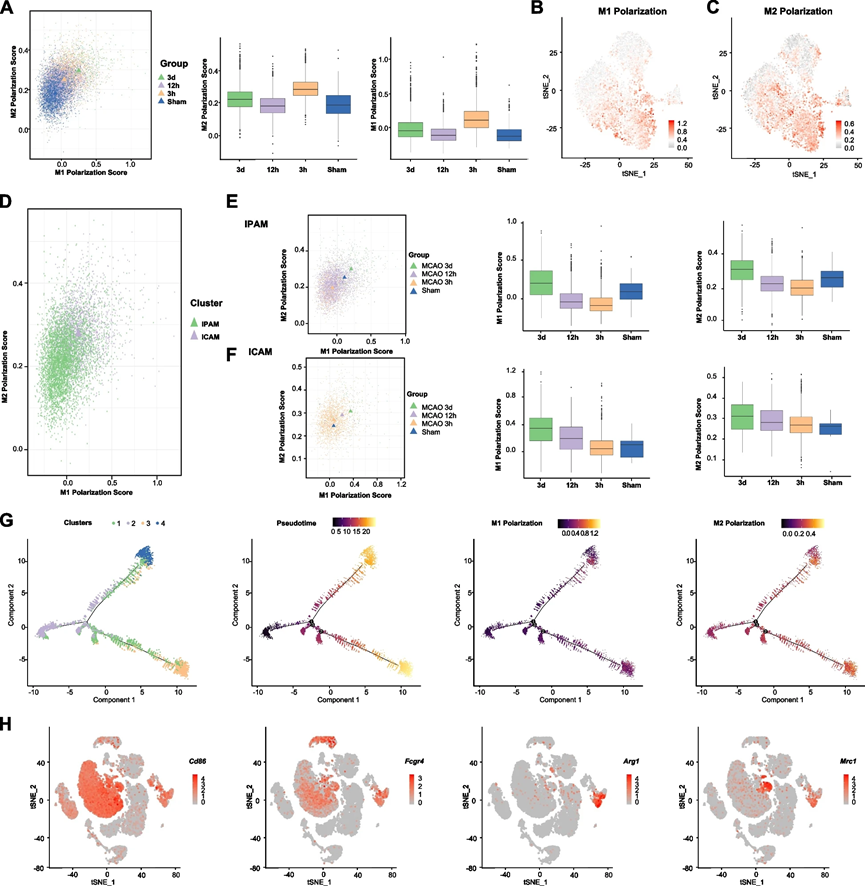
4、M1/M2 二分法無法對缺血后的小膠質細胞亞簇進行分類
在歷史上,基于體外刺激,微膠質細胞曾經簡單地被分類為促炎性的“M1”和抗炎性的“M2”。然而,根據我們的數據,M1極化評分的增加總是伴隨著不同時間點上升的M2極化評分(圖4A)。我們想知道我們的無監督聚類是否與這種表型分類相對應。為了測試這個假設,我們分別在IPAM和ICAM上執行了M1/M2極化評分。然而,ICAM的M1和M2極化評分都高于IPAM(圖4B-D)。此外,IPAM和ICAM在缺血損傷后的不同時間點上展現出相同的M1/M2極化改變趨勢(圖4E-F)。
接下來,我們還進行了偽時軌跡分析,并觀察到在缺血后出現了明顯的分歧軌跡。為了闡明這兩個分支是否與M1-和M2-表型微膠質細胞一致,我們檢查了不同軌跡中M1/M2標記基因的表達。在這兩個分支之間,M1/M2標記基因的表達仍然沒有顯著差異(圖4G)。基于許多關于M1/2微膠質細胞的已發表的體內研究,我們注意到研究人員使用Iba-1,這不僅在微膠質細胞中表達,而且在巨噬細胞中也表達,作為微膠質標記。他們還使用CD86和CD16(由Fcgr4編碼)標記“M1”微膠質細胞,使用Arg1和CD206(由Mrc1編碼)標記“M2”微膠質細胞。然而,根據我們的scRNA-seq數據,“M1”微膠質細胞標記物(Cd86和Fcgr4)在微膠質細胞和巨噬細胞中均有表達,而“M2”微膠質細胞標記物(Arg1和Mrc1)主要在巨噬細胞中表達,表明滲透的巨噬細胞可能被錯誤地認定為經典定義的“M2”微膠質細胞(圖4H)。因此,M1/M2的二分法可能無法識別不同的微膠質細胞亞群,并描繪它們在缺血后的功能。
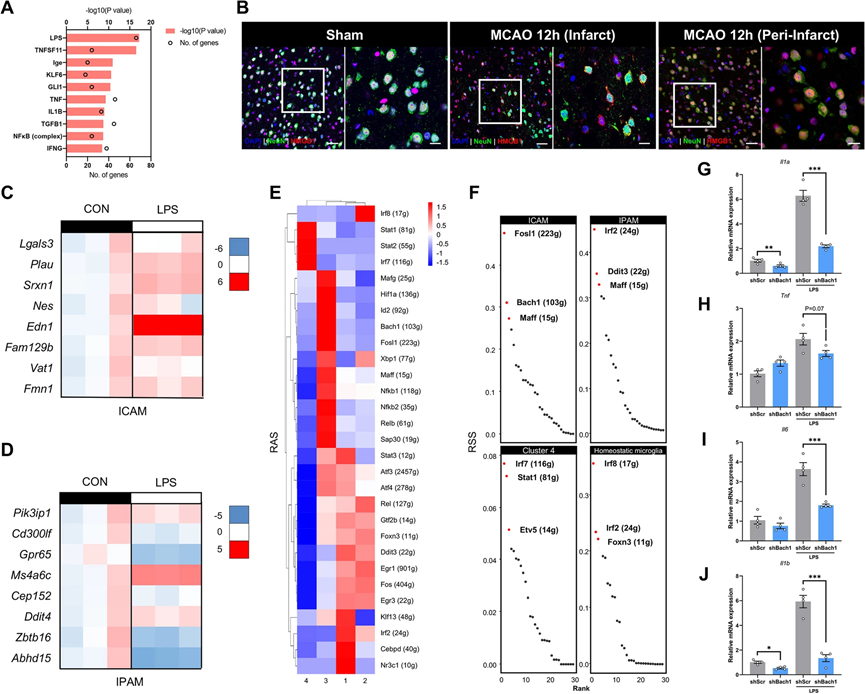
5、關鍵 DAMP 和調節子驅動 ICAM 生成
我們假設區域微環境信號可能驅動微膠質細胞代謝和功能的轉變,使其朝著ICAM和IPAM方向發展。通過使用Ingenuity System Pathway Analysis(IPA)軟件(QIAGEN),我們發現脂多糖(LPS)是推動ICAM生成的最重要的潛在因素之一(圖5A)。由于LPS是TLR4激活的經典配體,我們調查了在腦缺血后釋放的TLR4內源性配體HMGB1的空間分布。在MCAO后的12小時內,在梗死核內受損神經元內明顯出現HMGB1的胞質轉位,這表明HMGB1,作為一種被廣泛認識的損傷相關分子模式(DAMP),在梗死核釋放并可能驅動ICAM的形成(圖5B)。我們發現在LPS刺激后,大多數ICAM特異性標記基因在初級微膠質細胞中顯著上調,而IPAM標志基因的表達變化較小(圖5C,D)。為了調查垂死神經元釋放的DAMP是否誘導ICAM的生成,我們還通過多次凍融小鼠神經細胞系HT-22制備了神經源性的DAMP,如先前描述的 。結果表明,處理HT-22源DAMP的初級微膠質細胞顯著上調了ICAM標記基因,而IPAM標記沒有顯著差異。
除了細胞外的微環境信號外,我們還研究了哪些細胞內調控因子驅動ICAM的生成。使用單細胞調控網絡推斷和聚類(SCENIC)分析 來闡明缺血性卒中后微膠質細胞亞群形成的關鍵分子機制。通過全面考慮調控子活性評分(RAS)和調控子特異性評分(RSS),我們確定FOSL1和BACH1是最強大的ICAM特異性轉錄因子(TFs)之一(圖5E,F)。研究表明,在ST中,Fosl1在急性缺血后在缺血區域上調和富集。此外,BACH1下游靶基因的富集分析(使用Metascape)發現它們與炎癥顯著相關(例如,“細胞遷移的正調控”,“MAPK級聯的正調控”,“TNF-α NF-κB信號通路”,“細胞因子產生的調控”,“MyD88無依賴TLR4級聯”,“IL-17信號通路”,“對氧化應激的應答”和“模式識別受體信號通路”),這與ICAM的促炎作用一致。為了驗證BACH1對ICAM的調控能力,我們轉染了攜帶靶向Bach1的短發夾RNA(shRNA)的慢病毒的BV2細胞。Bach1-knockdown在LPS刺激后下調了ICAM特異性標記基因(Edn1,Plau)的表達水平,表明BACH1有潛力將穩態微膠質細胞轉化為ICAM。此外,LPS刺激后,經典的促炎因子,包括Tnf,Il1a,Il6和Il1b,在Bach1-knockdown細胞中也下調了(圖5G-J)。
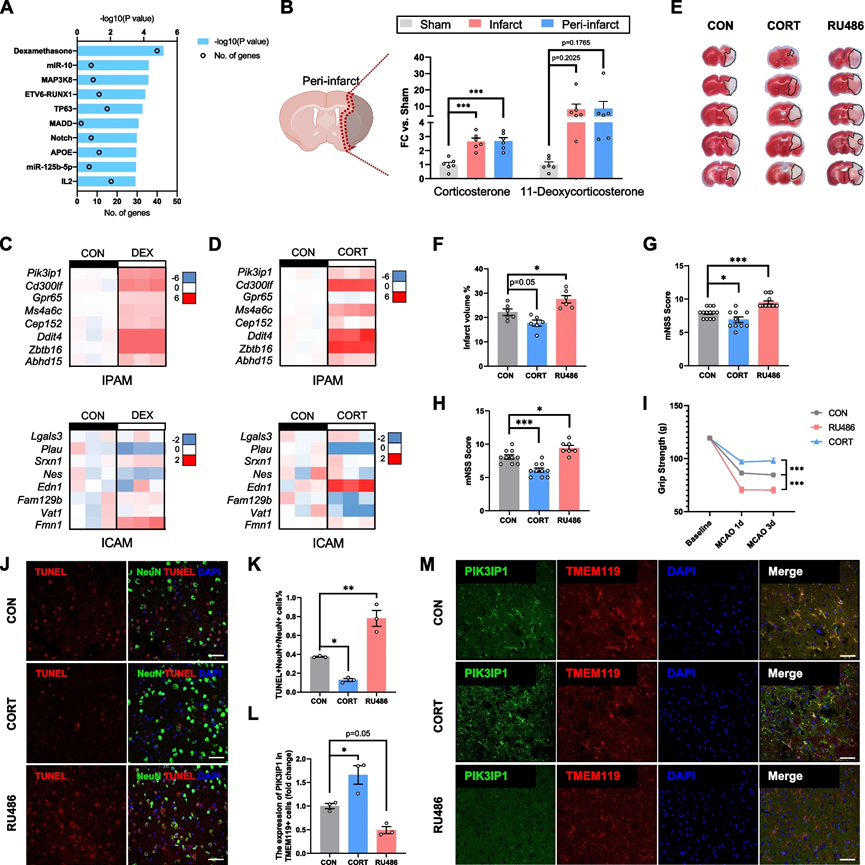
6、糖皮質激素觸發 IPAM 形成
為了識別IPAM的關鍵上游調控因子,使用了前200個IPAM標記基因進行IPA。令人驚訝的是,地塞米松(DEX)是誘導IPAM最強大的刺激因素(圖6A)。為了探索中風后糖皮質激素濃度的變化,我們進一步利用高通量靶向代謝組學來篩選在缺血后顯示變化的類固醇。根據定量數據,皮質酮和11-去氧皮質酮的水平在MCAO后在缺血核心和梗死周圍區域均顯著增加(圖6B)。為了驗證,我們使用DEX刺激初級微膠質細胞,強烈誘導了IPAM的生成,并顯著增強了IPAM特異性標記基因的表達(圖6C)。此外,小鼠主要的腎上腺皮質類固醇皮質酮也顯著上調了初級微膠質細胞中IPAM特異性標記基因的表達,表明糖皮質激素具有推動IPAM生成的強大能力(圖6D)。
為了在小鼠中驗證這些發現,對MCAO小鼠進行了皮質酮或糖皮質激素受體拮抗劑(RU486)的處理。令人驚訝的是,接受皮質酮處理的MCAO小鼠顯示出相對較輕的神經功能缺陷(圖6E-I)和減少的神經元凋亡(圖6J,K)。然而,在小鼠中RU486的投與導致更嚴重的神經功能缺陷(圖6E-I)和加劇的神經元死亡(圖6J,K)。
為了在不同實驗組中研究IPAM的變化,我們采用了免疫染色并測量了微膠質細胞中PIK3IP1的表達。與對照組相比,PIK3IP1的平均熒光強度在皮質酮處理組中顯著更高,而在RU486處理組中相對較低(圖6L,M)。因此,這些體外和體內的發現驗證了糖皮質激素在推動IPAM生成以及在缺血性卒中的急性階段中IPAM的神經保護性質中的決定性作用。
實驗方法:
慢病毒轉導、梗塞體積測量、神經行為測試、t-SNE 降維、scRNA-seq、GSVA、偽時間分析、軌跡推斷、SCENIC、TUNEL染色、空間轉錄組測序、實時定量 PCR、Western blot、RNAscope、靶向代謝組學。
參考文獻:
Li, H., Liu, P., Zhang, B. et al. Acute ischemia induces spatially and transcriptionally distinct microglial subclusters. Genome Med 15, 109 (2023).