






高分腫瘤文章的剖析——計算模擬+單細胞轉錄組學+機制

膠質母細胞瘤在其早期階段的進展仍然知之甚少。本研究中,作者在小鼠大腦中轉移PDGFB和genetic barcodes,以啟動膠質瘤發生,并能夠從最早的階段直接追蹤膠質母細胞瘤的進化。出乎意料的是,作者觀察到克隆消亡事件的高發生率和克隆大小的漸進性分化,甚至在惡性表型獲得后也是如此。計算模擬表明這些動力學是基于克隆的細胞-細胞競爭的結果。通過批量和單細胞轉錄組分析,結合譜系追蹤,作者揭示了Myc轉錄靶標與克隆大小不平衡的相關性最強。此外,作者還證明了下調Myc的表達足以驅動顱內移植膠質瘤中的競爭動力學。作者的研究結果提供了使用傳統回顧性方法無法獲得的膠質母細胞瘤進化的見解,突出了在該領域結合克隆追蹤和轉錄組學分析的潛力。本研究于2023年8月于《Cancer Cell》上發表,IF=50.3。
圖形摘要

技術路線

主要研究內容
1、膠質瘤誘導barcode library的驗證
為在體內誘導腫瘤啟動的基因組病變,同時對每個轉導的細胞進行特異標記,作者生成了一個致癌逆轉錄病毒載體的barcoded library。文庫中的每個載體包含:(i)一個獨特的genetic barcode,(ii)作為報告基因的GFP,(iii)PDGFB致癌基因(圖1)。作者將barcode設計為BH模式(一個non-A核苷酸后面接一個non-G核苷酸)的11次重復,最大理論復雜度為322(每個位置上可能的核苷酸數目上升到該位置上的數目),對應約3×1010形式。通過在低感染復數(0.01 MOI)下轉導5個獨立的NIH3T3細胞培養來測試所獲得的逆轉錄病毒文庫的有效復雜性和barcode分布的均勻性。隨后通過靶向NGS獲得的barcode組成分析顯示,其分布沒有重大偏差(圖1B,C)。庫(即,庫中功能向量中不同barcode的數量)的有效復雜度為(3±0.15)×104。同時,作者通過處理包含已知比例的兩個不同barcode克隆的細胞的不同樣本,驗證了作者的barcode檢索、測序和定量的方法。這項分析表明,即使在代表總樣本的10-4倍時,barcode也可以被一致檢索和量化。
研究將逆轉錄病毒文庫注射到E14小鼠胚胎的端腦囊泡中。正如已經報道的那樣,29個PDGFB的過表達在成年期早期總是導致膠質瘤的發展(圖1D)。隨著時間的推移,GFP表達標記的膠質瘤的總體質量(即GFP陽性細胞總數)增加(圖1E)。在早期出現癥狀的小鼠中,膠質瘤表現為小而彌漫的多灶性腫塊,類似于低級別腫瘤,而在后期出現癥狀的小鼠中,腫塊表現為更大和更密集的(圖1F-J)。這些數據與作者先前的觀察結果一致,即隨著時間的推移,膠質瘤從低級別進展到高級別,并伴隨著惡性程度的增加。
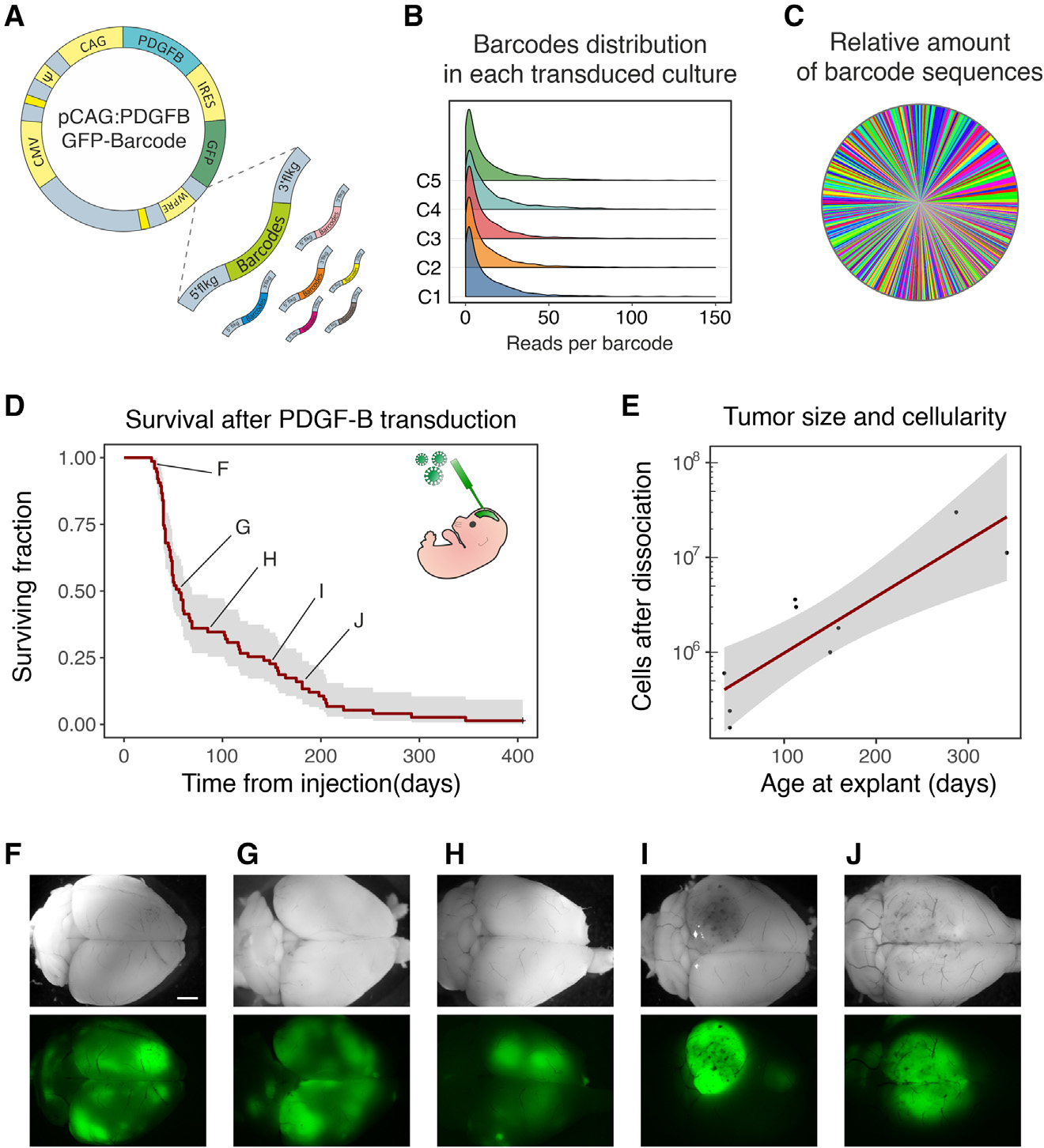
圖1 barcode矢量的構建和驗證
2、PDGFB誘導膠質瘤的克隆動力學
在注射(n=7)后第4天,從胚胎中提取DNA來估計逆轉錄病毒轉導細胞的數量。每個胚的barcode編號為(16±4)x 103。重要的是,在每個胚胎中,單個barcode的相對豐度是均一的,沒有過度代表的barcode,Gini不平等指數為0.51±0.02。該值與原始質粒文庫(0.57 ± 0.01)非常接近。隨后,對38只小鼠(16窩)在不同時間表現出神經癥狀的腫瘤塊提取DNA進行相同的分析。該分析顯示,發育過程中,在出生后151至342天的3只小鼠的主要克隆的數量急劇減少,從33日齡小鼠中的最多495個克隆到單個克隆(圖2A,C)。克隆數目的減少一方面與腫瘤質量和細胞數量的增加相呼應(圖1E-J),另一方面與它們相對分布的不平衡相呼應,如Shannon-Wiener熵指數(簡稱Shannon熵)所示,該指數隨時間呈指數下降趨向于0(圖2B)。這幅圖表明,隨著時間的推移,以犧牲其他克隆為代價而逐步占優勢的原始克隆的子集的選擇性擴張。
對這一現象的一個可能的解釋是少數細胞成功地克服了biological gate的發生,表現為在向完全惡性腫瘤進展過程中的克隆多樣性瓶頸。基于這一假設,晚期發病的膠質瘤,很可能已經通過了這一biological gate,如果移植到成年小鼠的大腦中,不應該面臨強大的克隆瓶頸。作者通過將5個遲發性膠質瘤(> 150天)的急性分離細胞移植到同系小鼠(每只膠質瘤至少2只小鼠)中,并比較原發性和繼發性膠質瘤的克隆組成,驗證了這一假設。移植動物在27 ~ 45天之間出現繼發性膠質瘤。隨后的克隆分析顯示,其中2例移植的膠質瘤是單克隆性的,因此,它們的繼發性腫瘤也是單克隆性的。另外3例移植瘤分別由2、3和5個克隆組成,但值得注意的是,每個克隆均產生單克隆繼發性腫瘤(圖2D)。盡管每個克隆的移植細胞數量(只有一個例外)遠高于產生繼發性腫瘤所需的最小數量,但這種完全的純化還是發生了(圖2E)。這表明,盡管有前期的進展,腫瘤細胞仍處于強烈的選擇性壓力之下。然而,這一結果可能受到親本無性系相對大小的強烈不平等的影響。為了解這種可能的限制,更好地理解在完全進展的腫瘤中正在進行的選擇,作者決定用只攜帶GFP和genetic barcodes的逆轉錄病毒庫標記完全進展的膠質瘤細胞后進行類似的移植實驗。作者使用了PDGFB誘導的膠質瘤(CSC-204D39)的原代培養,已知當原位移植到同系小鼠(穿透率> 97 %,n = 64)時,通常會產生繼發性腫瘤。將文庫轉染的細胞移植到成年小鼠(n = 4)的大腦中,當小鼠出現神經癥狀時收集二次腫瘤。對移植前的細胞和來自二次腫瘤塊的細胞進行barcode測序。有趣的是,腫瘤的二次生長伴隨著barcode數量的急劇減少,barcode數量從(1.19 ± 0.05)× 104減少到1(n = 4,圖2D),這表明即使在進展的膠質瘤中也發生了類似于在膠質瘤進展過程中觀察到的瓶頸。這一結果強烈表明,至少一些負責克隆減少的機制可能在獲得完全惡性腫瘤后仍在運作,指出存在進一步的腫瘤內選擇來源。
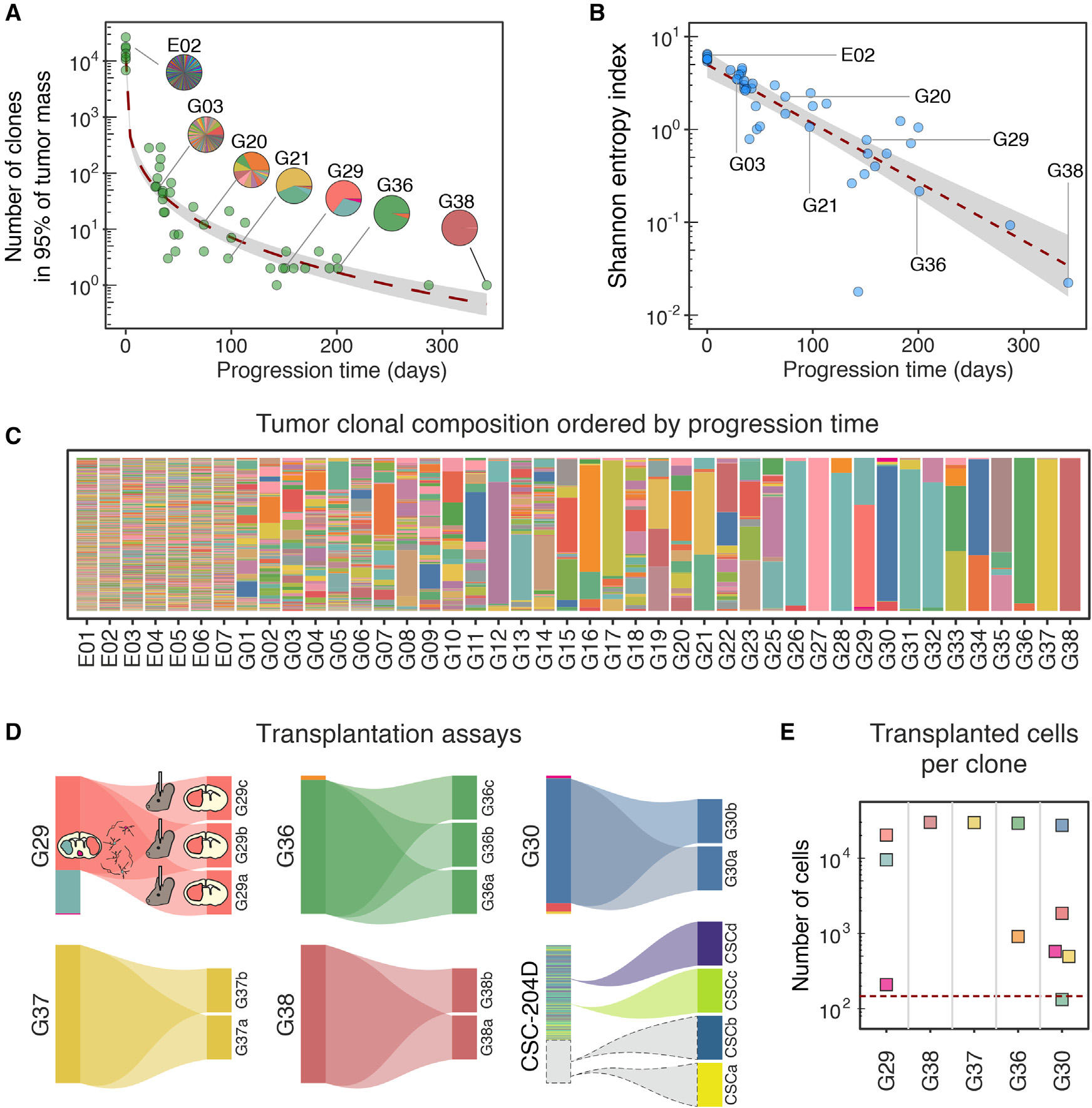
圖2 原發性和繼發性膠質瘤的克隆組成動態
3、膠質瘤如何失去克隆異質性?
在作者的膠質瘤發生模型中觀察到的克隆減少,理論上可以歸因于幾種不同的非互斥機制:(i)高比率細胞死亡(或細胞周期撤退)導致的統計漂移。細胞死亡或靜息可以隨機驅動克隆不平衡,從而降低克隆異質性。(ii)細胞周期長度的克隆差異。如果克隆顯示不同的細胞周期長度,較快克隆的大小將以指數方式偏離較慢克隆的大小,它們最終將在腫瘤團塊中占優勢,使其他克隆過度生長,并減少明顯的異質性。(iii)基于克隆身份的細胞-細胞競爭。在器官發育和腫瘤發生過程中,特定基因產物表達或活性的遺傳性(即,克隆性)差異會引發相鄰細胞之間的競爭,為克隆選擇提供基礎。為研究這些機制對克隆多樣性減少的可能影響,作者進行了Monte Carlo simulations,其中相關參數可以單獨操縱,以探索它們在塑造克隆動態中的作用。在作者的模擬中,一個“virtual tumor cells”的起始集在一個簡化的膠質瘤生長模型上,該模型是由在體外或體內進行的實驗措施所產生的現實值。作者開始使用這個框架來檢驗細胞死亡對克隆多樣性降低的影響。首先,作者考慮了一個極限場景,其中所有細胞的周期設置為體外測得的平均長度(22 h),并且在整個模擬過程中細胞死亡概率(pCD)是恒定的。這里作者注意到,當pCD達到一定值時,細胞死亡克服增殖,模擬種群數量減少,最終消失。超過180天,pCD的最高值仍然與腫瘤生長(即每小時約有3 %的死亡細胞)相容,導致克隆多樣性從10000個下降到580個克隆(圖3A)。與體內(大約4個數量級)相比,這種降低小于1.5個數量級,幾乎可以忽略不計。此外,這種細胞死亡率遠遠高于TUNEL分析所觀察到的結果,在體內中,死亡細胞的比例不到0.01 %(圖3B)。作者隨后引入了一個不同的細胞死亡模型來考慮腫瘤負荷的影響。因此,細胞死亡的概率被建模為腫瘤細胞總數的邏輯函數。作者通過探索nCell50參數來研究不同腫瘤負荷強度的影響。該參數對應于負責一半腫瘤負荷效應的細胞數(即Logistic曲線的拐點)。即使在這種情況下,細胞死亡要么太明顯,不允許腫瘤生長,要么太輕微,不能重復實驗觀察到的克隆多樣性減少的程度(圖3C)。同樣,在這種情況下,可檢測到的克隆減少(每小時約有3.2%的死亡細胞)所對應的細胞死亡凈百分比與作者的TUNEL分析結果不一致。上述結果排除了在膠質瘤發生過程中高細胞死亡率可以自行解釋觀察到的克隆動態的可能性。
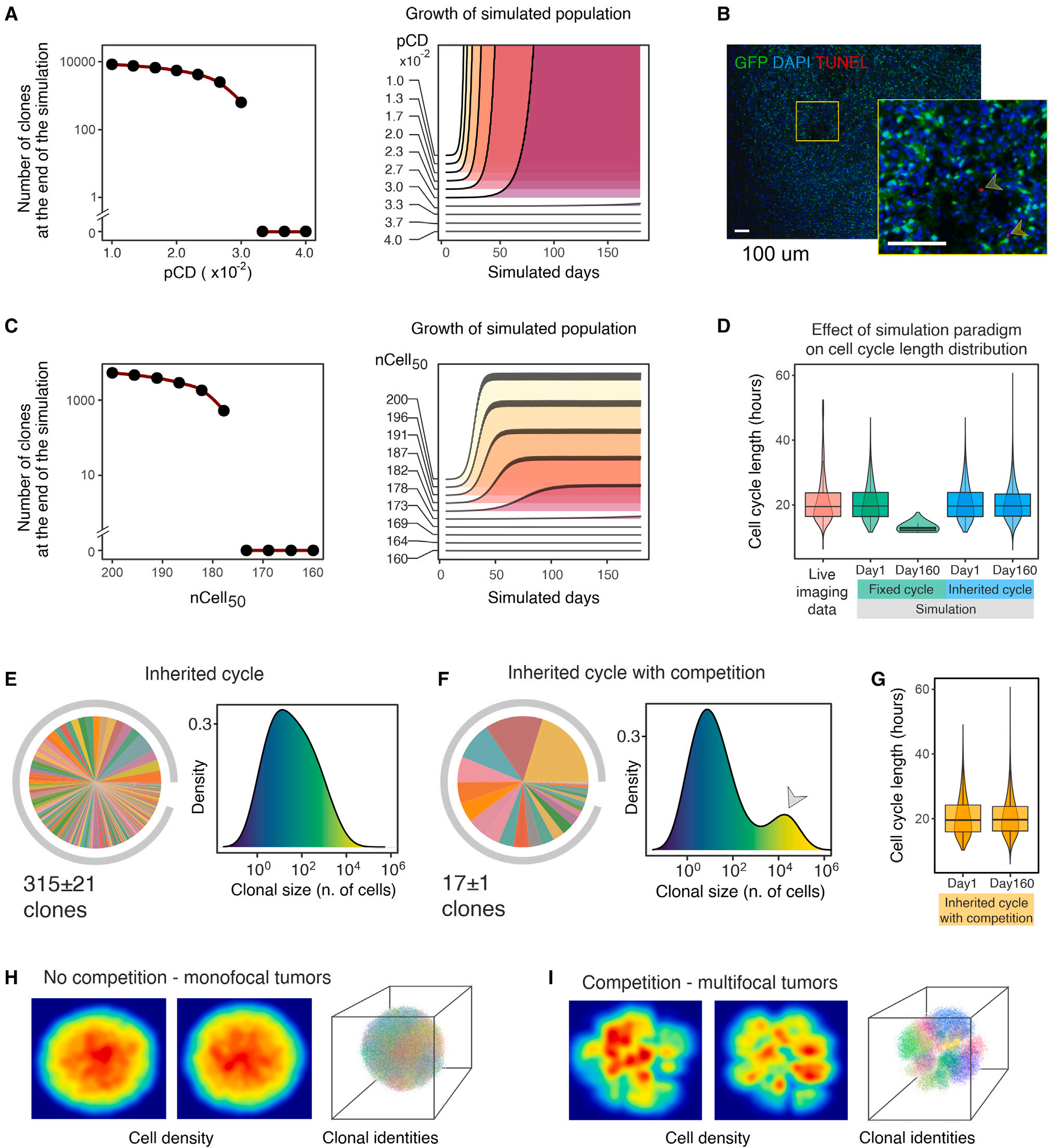
圖3 膠質瘤生長的計算分析
4、局部細胞密度和細胞周期長度波動
接下來,作者增加了模型的復雜性,包括細胞周期長度的克隆差異。作者從理論上的極端假設出發,即每個子細胞從其母細胞繼承一個固定的、未被修飾的細胞周期長度。在此假設下,通過初始化由視頻延時顯微鏡測量的細胞周期長度分布的原始群體,模擬結果產生了與作者在體實驗數據不兼容的圖片。更快的循環細胞的選擇性擴張導致了中位細胞周期長度隨時間的下降,下降到約14 h,這比實驗觀察到的明顯縮短(圖3D)。盡管這種不切實際的行為,但在160個模擬日中,克隆的數量從10,000個變化到約200,比作者所觀察到的減少得更少。更真實的模擬包括細胞分裂后細胞周期長度的變化。在這種范例中,細胞繼承其父代的細胞周期長度,加上一個正態分布的隨機值。這種模擬結果使細胞周期長度的分布更接近實驗數據(圖3D)。在此條件下,同樣地,雖然幾個生物學參數正確地再現了(如總體腫瘤生長率,細胞周期長度和腫瘤體積的分布),但在160個模擬日中,克隆數量從10000個下降到約665個(圖3E)。再次,該結果與實驗數據不符。因此,克隆多樣性降低似乎不太可能僅僅是由于細胞周期長度差異造成的。
5、細胞與細胞之間的競爭
最后,作者在上述模型中加入了基于克隆身份的細胞-細胞競爭的貢獻。通過區分同胞(同一克隆)和無關(不同的克隆)細胞,每個細胞的適合度受到其鄰近細胞的影響,這取決于它們各自的克隆身份。特別地,作者建模了一個場景,對于任意給定的細胞,不相關的鄰居比兄弟姐妹對細胞死亡概率的影響更大。在這個模擬中,作者獲得了從10000到108的減少,值得注意的是,其中只有17 ± 1占總人口的95 %,Shannon熵為2.5 ± 0.1。此外,由于超級競爭者克隆的出現,克隆大小呈現多峰分布(圖3F)。值得注意的是,觸發局部克隆競爭并不影響細胞周期長度分布(圖3G),并導致模擬的腫瘤腫塊具有與實際體內腫瘤大體相似的大小和拓撲結構,顯示出密集的分葉被幾乎無瘤的區域(圖3H,I)隔開。沒有其他經過檢驗的假設導致如此明顯的克隆不平衡和多樣性減少。
6、進展中的腫瘤細胞動力學(如何競爭)
為深入了解細胞-細胞競爭的可能機制,作者對PDGFB誘導的不同時間收獲的膠質瘤進行了RNA測序(RNA sequencing, RNA-seq)分析,(n = 4只小鼠,出生后39天)之前或神經癥狀發作時(n = 7只小鼠,出生后40 ~ 176天)。由于該barcode序列在結構上與高表達的PDGFB-IRES-GFP轉錄本相連,因此可以通過RNA-seq分析高效檢索。這使得作者可以直接估計每個腫瘤的Shannon熵,并將其與性狀相關基因(trait-associated gene,TAG)分析中的基因表達相關聯。然后使用TAG衍生的排序來驅動基因集富集分析(GSEA),在Hallmark基因集上進行,克隆復雜性的降低與Myc信號直接相關(Myc Targets V1和E2F靶點緊隨Myc靶點V2,p < 10-5,圖4B,C)。這個結果是非常有意義的,因為Myc信號傳導是涉及細胞-細胞競爭的少數已描述的機制之一。Myc表達本身是超級競爭者克隆的特征,與Shannon熵顯著負相關,這表明Myc作為克隆丟失(Pearson相關性: 0.85,p < 0.001,圖4D)的驅動因子的潛在作用。作者推測,Myc水平的升高可能是由于對表達較高水平Myc的細胞進行選擇,從而逐漸富集的結果。然而,考慮到在完全進展的腫瘤中也有在單個克隆中持續選擇的跡象(圖2D),作者想知道即使完全純化,單克隆腫瘤是否會在細胞水平上保持異質性的Myc表達。為解決這個問題,作者分離了兩個單克隆腫瘤,并通過基線稀釋克隆分離亞克隆培養物。然后,作者量化了每個腫瘤10個亞克隆的Myc表達,并將差異與親代培養中觀察到的波動進行了比較。在這兩種腫瘤中,作者觀察到亞克隆培養物中Myc表達的異質性高于親代腫瘤(圖4E)。作者利用RNA-seq數據佐證了這一證據,以提示在膠質瘤進展過程中克隆內異質性的維持。在膠質瘤進展的早期階段(PDGFB轉導后小于45天)中,大多數可檢測到的突變并不是在所有的膠質瘤細胞(等位基因頻率中位數= 0.20)中共享的。在后期,出現了一組廣泛的突變(等位基因頻率中位數= 0.93),可能是某些克隆擴張的跡象,與作者之前的數據一致。盡管如此,低頻突變仍然是絕大多數,這表明基因組變異在擴大的克隆中不斷涌現(圖4F)。
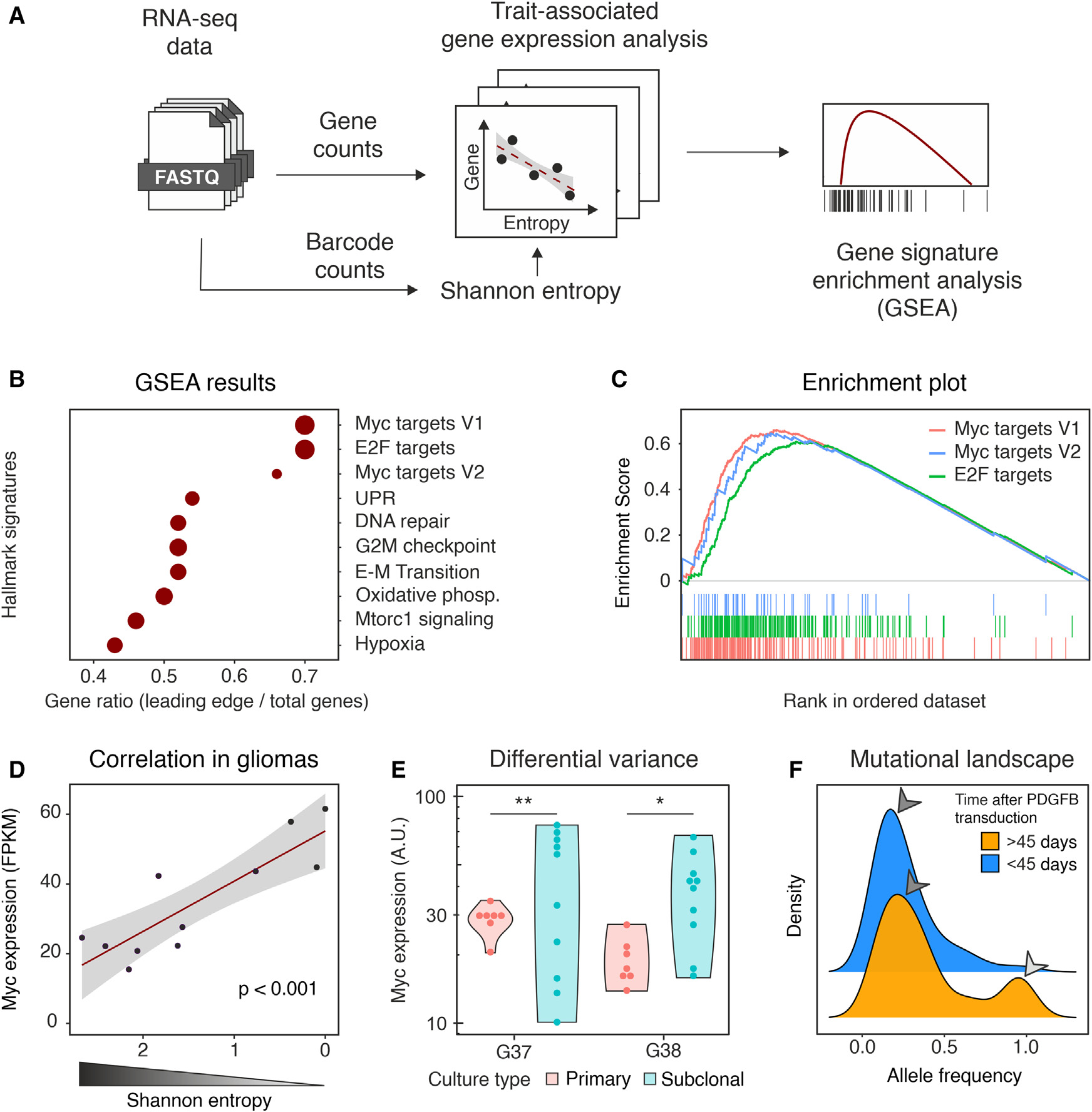
圖4 與進展性膠質瘤bulk RNA-seq相關的克隆分析
7、膠質瘤的早期克隆分化
為對克隆間競爭的潛在驅動因素進行更高分辨率的搜索,作者對膠質瘤進行了單細胞RNA-seq(sc RNA-seq)分析,重點關注可能存在許多不同克隆且其大小開始發散的階段。根據作者之前的分析,這對應于出生后大約40天(圖2A)。分離3個獨立的PDGFB誘導的膠質瘤細胞,通過熒光激活細胞分選分離出GFP陽性的細胞組分,并用Chromium 10X進行單細胞分選。同時,通過對barcode區域的靶向DNA測序,分析了分離細胞的代表性部分(5 %)。后一種分析使作者能夠確定每個barcode克隆的大小。然后,在scRNA-seq數據中搜索本分析中發現的barcode,將細胞的轉錄組圖譜與克隆信息聯系起來,并將其與屬于的克隆的大小和身份聯系起來(圖5A-5C)。數據顯示,克隆通常由屬于不同轉錄簇的細胞組成(圖5D)。值得注意的是,在GSEA分析中,當比較屬于小克隆(單個細胞占腫瘤質量的30%以下,累計約占測序細胞總數的二分之一)的細胞和屬于大克隆(分別代表至少30 %的腫瘤塊,并累積組成另一半測序細胞)的細胞時,最富集的基因簽名是Myc信號和相關的E2F-靶標,再次是(p < 10-4,見圖5E、F)。這增加了Myc-E2F軸在控制克隆大小方面的作用的證據,并指出其在膠質瘤發生過程中塑造細胞-細胞競爭動力學的重要性。進一步分析顯示,不同大小克隆的細胞中轉導的PDGFB轉基因表達沒有差異,而大克隆的細胞中Myc表達有較高的趨勢(圖5G)。
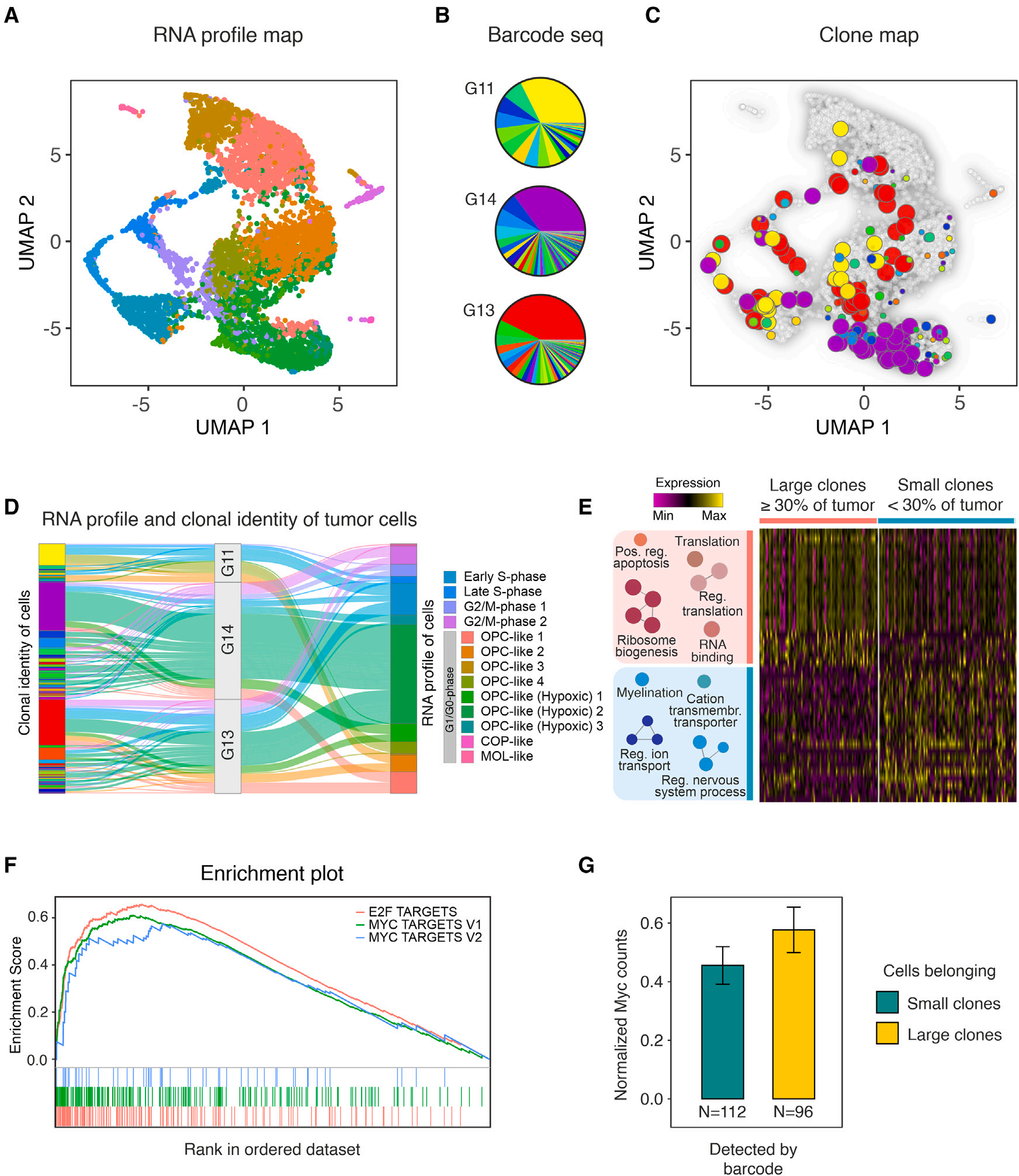
圖5 早期膠質瘤的單細胞RNA-seq分析
8、膠質瘤細胞中Myc介導的競爭
然后,作者從功能上評估了Myc表達水平在基于克隆的競爭中的作用。作者使用CSC-204D細胞,即PDGFB驅動的膠質瘤原代培養細胞,用于圖2D所示的實驗。作者通過穩定轉導兩種不同的攜帶GFP的逆轉錄病毒載體和一種旨在抑制Myc表達的人工miRNA(amiRNA)來構建CSC-204D細胞。作者通過qPCR驗證了兩種不同的anti-Myc amiRNAs(miRMyc16和miRMyc19)介導的敲低,分別降低到原來的52 ± 4%和61 ± 6%(圖6A)。這種調節水平與作者在原代膠質瘤細胞中觀察到的情況非常相似(圖4D)。隨后,作者通過MTT實驗比較了Myc敲低(Myc-knockdown,Myc-KD)細胞和轉染scramble miRNA的細胞的體外生長速率,培養物的復制時間無明顯變化(圖6B,C)。作者進行了移植實驗以測試Myc-KD膠質瘤細胞單獨移植或與Myc野生型(Myc-wt)細胞混合移植時的行為。特別地,作者設計了兩個互補的實驗來推斷Myc在膠質瘤生長和克隆動力學中的作用:在第一個實驗中(實驗1),作者將Myc-KD細胞(用RFP和GFP標記)單獨原位移植到小鼠大腦中,并檢查它們是否可以作為它們的親本群體產生繼發性膠質瘤。在第二個實驗(實驗2)中,作者原位移植相同數量的Myc-KD細胞與等數量的Myc-wt細胞(僅用RFP標記)混合,檢測這兩個亞群是否可以共同生長,或者Myc-KD細胞在繼發性膠質瘤中是否會被淘汰。作者推斷,實驗1中膠質瘤的發展,以及實驗2中Myc-wt細胞的純化趨勢,表明Myc在膠質瘤進展過程中發揮了驅動競爭的作用(圖6D)。事實上,實驗1中所有移植的小鼠都發生了繼發性膠質瘤(7/7)。值得注意的是,膠質瘤的發生時間與實驗2相同,Myc-KD和Myc-wt膠質瘤細胞在體內的成瘤能力和生長速度沒有差異(圖6E-6G和6J)。在實驗2中,所有的移植小鼠(7/7)都發生了繼發性膠質瘤,并且Myc-wt細胞被高度純化。幾乎沒有Myc-KD細胞(RFP和GFP雙標記)被發現(圖6H-K)。在作者使用相同細胞進行的體外實驗中也得到了類似的結果。作者將Myc-KD(或scrambled miRNA control)細胞和Myc-wt細胞以1:1的比例混合在U型多孔板中形成球體。然后作者在三個時間點監測他們的生長情況。
Myc-KD細胞被Myc-wt細胞取代的比率顯著高于對照膠質瘤細胞(圖6L-N)。總之,這些結果證實了作者提出的Myc在膠質瘤發生過程中驅動基于克隆的競爭的作用。
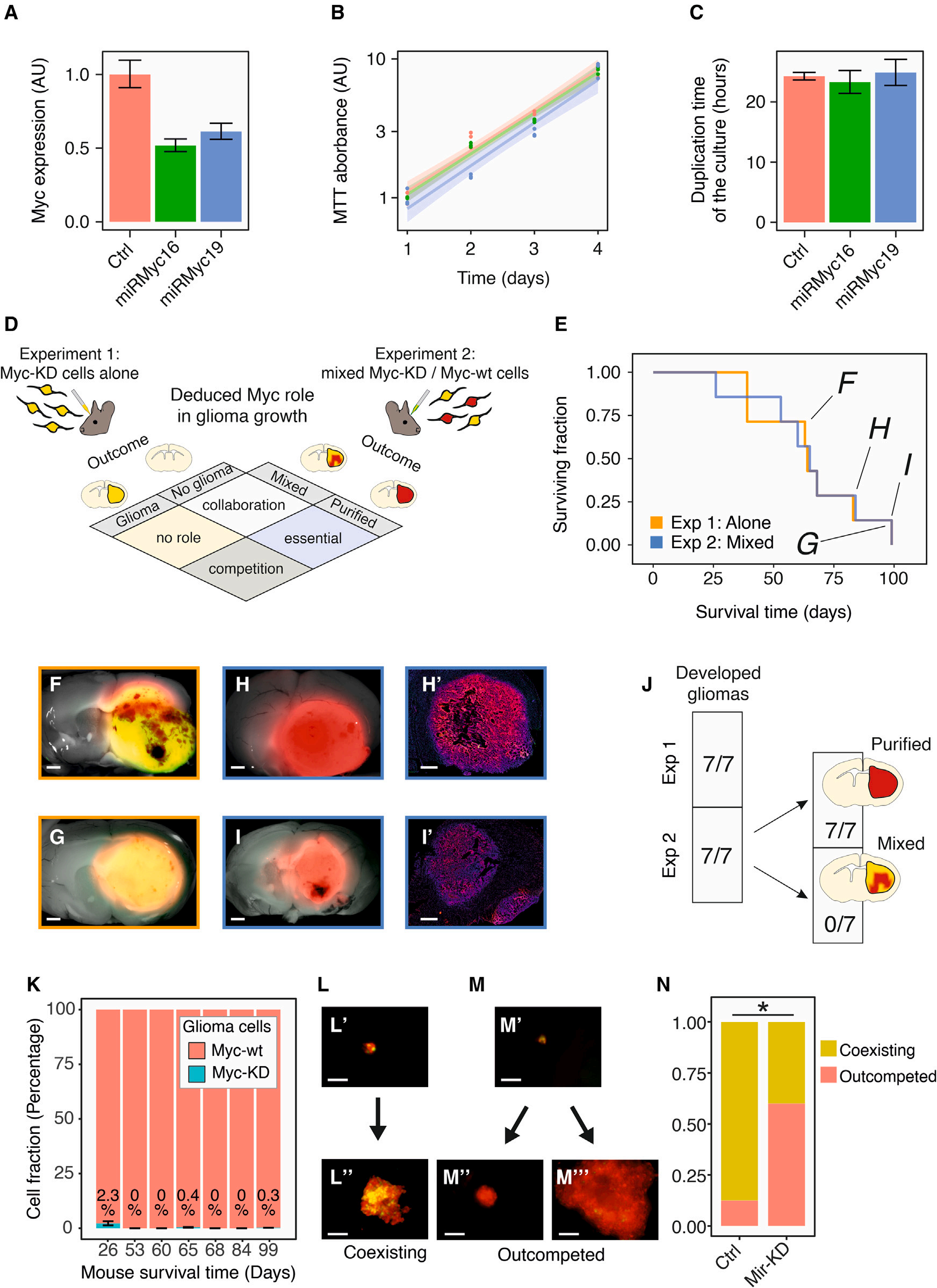
圖6 Myc modulation驅動膠質瘤生長的競爭動態
實驗方法
C57BL/6J小鼠,膠質瘤細胞原代培養,膠質瘤細胞的極限稀釋克隆,膠質瘤細胞中Myc下調,免疫染色和TUNEL試驗,定量PCR,逆轉錄病毒barcoding libraries production,Barcode sequencing,Bulk RNA sequencing,scRNA-seq,基因表達分析,基因組變異分析
參考文獻
Ceresa D, Alessandrini F, Lucchini S, Marubbi D, Piaggio F, Mena Vera JM, Ceccherini I, Reverberi D, Appolloni I, Malatesta P. Early clonal extinction in glioblastoma progression revealed by genetic barcoding. Cancer Cell. 2023 Aug 14;41(8):1466-1479.e9. doi: 10.1016/j.ccell.2023.07.001. Epub 2023 Aug 3. PMID: 37541243.